XiaoMi-AI文件搜索系统
World File Search SystemBinning

GSPRINT5514BSI 产品传单
GSPRINT5514BSI 具有 4608 x 3072 像素,每个像素为 5.5 µm 见方 - 4/3 宽高比 4k 传感器,与 APS-C 光学元件兼容。GSPRINT5514BSI 具有 10 位输出,可实现每秒 670 帧。在 12 位模式下,传感器输出 350 fps。使用背面照明技术,对于 UV 应用,传感器在 510 nm 处实现 86% 的量子效率,在 200 nm 处实现 17% 的量子效率。该传感器提供双增益 HDR 读出,最大限度提高 15 ke- 满阱容量,最小 < 2.0 e- 噪声,实现出色的 78.3 dB 动态范围。模拟 1x2 合并将满阱容量增加到 30 ke-。图像数据通过 84 个 sub-LVDS 通道以 1.2 Gbps 的速度输出。对于不需要最大帧速率的应用,可以使用多路复用模式将输出通道数减少 2 的任意倍数。GSPRINT5514BSI 有单色或彩色版本,配有密封或可拆卸盖玻片,并采用 454 针 µPGA 封装。

FEMA:针对大样本全脑成像数据的快速高效混合效应算法
图 1 分箱对固定效应参数估计的均方误差 (MSE) 的影响。我们模拟了 2000 个具有 5 个固定效应(10,000 个观测值)的成像变量。然后,使用 20 个不同的箱值,我们使用 FEMA 估计参数并计算参数估计的平均(超过 50 次重复)平方误差。面板 (a) 中的黑色虚线表示五个固定效应中的每一个的总 MSE(跨 2000 个成像变量),而橙色实线表示五个固定效应的总 MSE 的平均值。我们观察到最小总 MSE 在箱值为 100(由绿线表示)时,而箱值为 20(由紫线表示)显示出可比的 MSE;面板 (b) 显示每个箱值所需的计算时间(跨 50 次重复取平均值);箱值为 20(紫线)的计算时间是箱值为 100(绿线)所需计算时间的一小部分。请注意,两个面板的 x 轴都是非线性的。

硅光子晶体腔阵列的转移印刷微组装:超越制造公差极限
光子晶体腔 (PhCC) 可以将光场限制在极小的体积内,从而实现高效的光物质相互作用,以实现量子和非线性光学、传感和全光信号处理。微制造平台固有的纳米公差可能导致腔谐振波长偏移比腔线宽大两个数量级,从而无法制造名义上相同的设备阵列。我们通过将 PhCC 制造为可释放像素来解决此设备可变性问题,这些像素可以从其原生基板转移到接收器,在接收器中有序的微组装可以克服固有的制造差异。我们在一次会话中演示了 119 个 PhCC 中的 20 个的测量、分箱和传输,产生了空间有序的 PhCC 阵列,21 按共振波长排序。此外,设备的快速原位测量首次实现了 PhCC 对打印过程的动态响应的测量,在几秒到 24 小时的范围内显示出塑性和弹性效应。25

利用迭代正则化流进行相空间均匀数据选择
计算和实验能力的提高正在迅速增加日常生成的科学数据量。在受内存和计算强度限制的应用中,过大的数据集可能会阻碍科学发现,因此数据缩减成为数据驱动方法的关键组成部分。数据集在两个方向上增长:数据点的数量和维数。降维通常旨在在低维空间中描述每个数据样本,而这里的重点是减少数据点的数量。提出了一种选择数据点的策略,使它们均匀地跨越数据的相空间。所提出的算法依赖于估计数据的概率图并使用它来构建接受概率。当仅使用数据集的一小部分来构建概率图时,使用迭代方法来准确估计稀有数据点的概率。不是对相空间进行分组来估计概率图,而是用正则化流来近似其函数形式。因此,该方法自然可以扩展到高维数据集。所提出的框架被证明是在拥有大量数据时实现数据高效机器学习的可行途径。
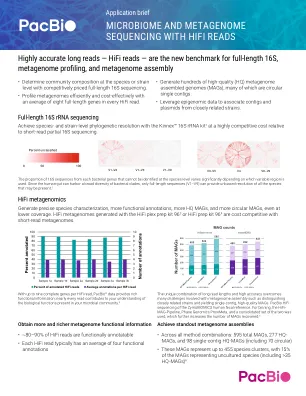
应用简介-使用 HiFi 进行宏基因组测序-...
1. 应用说明 – Kinnex 16S rRNA 试剂盒用于全长 16S 测序 2. Johnson, JS 等人 (2019) 评估 16S rRNA 基因测序在物种和菌株水平微生物组分析中的应用。《自然通讯》。10(1),5029。 3. 程序和清单 – 使用 HiFi plex 制备试剂盒 96 制备多重全基因组和扩增子文库 4. 程序和清单 – 使用 HiFi 制备试剂盒 96 制备全基因组文库 5. Gehrig, J. 等人 (2022) 找到合适的选择:评估短读和长读测序方法以最大限度提高临床微生物组数据的效用。《微生物基因组学》,8(3),10.1099/mgen.0.000794。 6. Portik, DM 等人(2024) 使用长读组装、分箱和合并方法从人类肠道微生物群中高度准确地组装宏基因组。bioRxiv。doi:https://doi.org/10.1101/2024.05.10.593587 7. 概述 – HiFi 应用选项和测序建议。8. 程序和清单 – 使用条形码引物扩增细菌全长 16S 基因。9. 程序和清单 – 从 16S rRNA 扩增子制备 Kinnex 文库

新的长读元基因组装方法增加了宿主和环境微生物组的高质量磁质量
1。冯等人。2022。高保真长读的元基因组组装,用hifiasm-meta读取。自然方法,19:671–674。2。Benoit等。2024。使用MetAMDBG的长期准确读取的高质量元基因组组件。 自然生物技术,https://doi.org/10.1038/s41587-023-01983-6 3。 Chklovski等。 2023。 checkm2:一种使用机器学习评估微生物基因组质量的快速,可扩展和准确的工具。 Biorxiv,https://doi.org/10.1101/2022.07.11.499243 4。 Kang等。 2019。 metabat 2:一种自适应分解算法,用于元基因组组件的稳健有效基因组重建。 peerj,7:e7359。 5。 Pan等。 2023。 semibin2:自我监督的对比学习可以为短而长阅读的测序提供更好的磁磁。 生物信息学,39:I21 – I29。 6。 Sieber等。 2018。 通过消除,聚合和评分策略从宏基因组中恢复基因组。 自然微生物学,3:836–843。 7。 Chaumeil等。 2019。 GTDB-TK:一种将基因组与基因组分类学数据库进行分类的工具包。 生物信息学,35:1925-1927。使用MetAMDBG的长期准确读取的高质量元基因组组件。自然生物技术,https://doi.org/10.1038/s41587-023-01983-6 3。Chklovski等。2023。checkm2:一种使用机器学习评估微生物基因组质量的快速,可扩展和准确的工具。Biorxiv,https://doi.org/10.1101/2022.07.11.499243 4。Kang等。 2019。 metabat 2:一种自适应分解算法,用于元基因组组件的稳健有效基因组重建。 peerj,7:e7359。 5。 Pan等。 2023。 semibin2:自我监督的对比学习可以为短而长阅读的测序提供更好的磁磁。 生物信息学,39:I21 – I29。 6。 Sieber等。 2018。 通过消除,聚合和评分策略从宏基因组中恢复基因组。 自然微生物学,3:836–843。 7。 Chaumeil等。 2019。 GTDB-TK:一种将基因组与基因组分类学数据库进行分类的工具包。 生物信息学,35:1925-1927。Kang等。2019。metabat 2:一种自适应分解算法,用于元基因组组件的稳健有效基因组重建。peerj,7:e7359。5。Pan等。2023。semibin2:自我监督的对比学习可以为短而长阅读的测序提供更好的磁磁。生物信息学,39:I21 – I29。6。Sieber等。 2018。 通过消除,聚合和评分策略从宏基因组中恢复基因组。 自然微生物学,3:836–843。 7。 Chaumeil等。 2019。 GTDB-TK:一种将基因组与基因组分类学数据库进行分类的工具包。 生物信息学,35:1925-1927。Sieber等。2018。通过消除,聚合和评分策略从宏基因组中恢复基因组。自然微生物学,3:836–843。7。Chaumeil等。 2019。 GTDB-TK:一种将基因组与基因组分类学数据库进行分类的工具包。 生物信息学,35:1925-1927。Chaumeil等。2019。GTDB-TK:一种将基因组与基因组分类学数据库进行分类的工具包。生物信息学,35:1925-1927。

宏基因组学深度学习方法:评论
于2023年12月20日收到; 2024年3月27日接受; 2024年4月17日出版作者分支:1 IRD,索邦大学,Ummisco,32 Avenue Henry Varagnat,Bondy Cedex,法国; 2 Sorbonne University,Inserm,Nutriomics,91 BVD de L'Hopital,法国75013,法国。*信函:加斯帕·罗伊(Gaspar Roy),加斯帕(Gaspar。 Jean-Daniel Zucker,Jean-Daniel。Zucker@ird。FR关键字:微生物组;宏基因组学;深度学习;神经网络;嵌入; binning;疾病预测。缩写:ASV,扩增子序列变体; CAE,卷积自动编码器; CGAN,有条件的生成对抗网络; CNN,卷积神经网络; Dae,Denoing AutoCododer; DL,深度学习; FFN,馈送网络; GAN,生成对抗网络;它的内部转录垫片; LSTM,长期记忆; MAG,元基因组组装基因组; MGS,宏基因组; MIL,多个实例学习; ML,机器学习; MLP,多层感知器; NGS,下一代测序; NLP,自然语言处理; NN,神经网络; RNN,经常性神经网络; SAE,稀疏的自动编码器; Sota,艺术状态; SVM,支持向量机; TNF,四核苷酸频率; Vae,各种自动编码器; WGS,全基因组测序。数据语句:文章或通过补充数据文件中提供了所有支持数据,代码和协议。补充材料可与本文的在线版本一起使用。001231©2024作者

IDE161 是一种潜在的首创临床候选 PARG 抑制剂,选择性靶向具有复制压力和 DNA 修复脆弱性的实体肿瘤
A) 描绘了对 TOP1 和 PARG 双重抑制的拟议 MOA 的模型。B) 对 PRISM 化合物和 PARGi 的反应的 Spearman 相关图;橙色表示 TOP1 抑制剂,黑色表示其他。(插图)按 MOA 分组的顶级相关化合物的 Swarmplot(未显示少于 2 种化合物的 MOA)。拓扑异构酶抑制剂 (TOP)、法呢基转移酶 (FT)、微管蛋白聚合 (TP)、极光激酶 (AK)、胸苷酸合酶 (TS)。C) 使用 PAR MSD 测定法评估 PAR 链积累。值绘制为相对于 DMSO 对照的平均值 ± SD。使用 Student's t 检验进行统计分析;ns(不显著)、**(<0.01)、***(<0.001)。D)(左)使用基于抗 TOP1cc 抗体的免疫荧光测定法在指示时间点测量 TOP1-DNA 裂解复合物 (TOP1cc)。根据单个细胞中的 TOP1cc 平均强度值进行群体分箱和非线性曲线拟合。使用 Kruskal-Wallis 检验进行统计分析;****(<0.0001)。(右)使用基于抗 γ -H2AX 抗体的免疫荧光测定法检测核 γ -H2AX。值(平均值 ± SD)绘制为 γ -H2AX 平均强度范围的百分比群体。E)从 CldU 标记的 DNA 纤维测量结果显示,IDE161 和 CPT 介导的复制叉减慢。框表示中位数和 IQR。使用 Mann-Whitney U 检验进行统计分析;*(<0.05),**** (<0.0001)。
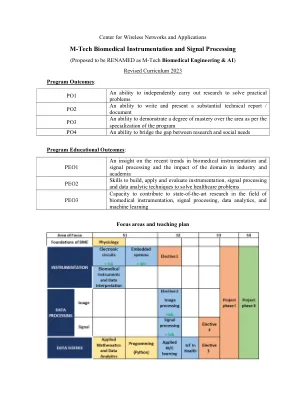
M-Tech 生物医学仪器和信号处理
CO1 能够理解数据挖掘过程中涉及的步骤(例如预处理、分类、回归、聚类和可视化)并将其应用于医疗数据的分析。 CO2 能够描述不同的预测分析方法及其在医疗领域的应用。 CO3 能够评估来自不同来源的数据以创建有意义的演示文稿。 课程内容 使用 Python 进行数据分析:了解数据 - (a)属性、数据的统计描述、数据可视化、相似性 - 不相似性、(b)预处理 - 缺失值、噪声数据、数据缩减、数据转换 - 规范化、标准化、分箱、聚类。 使用 Python 进行应用数学:数学基础 - 线性代数 - 向量、矩阵、特征值、特征向量、奇异值分解、降维、主成分分析、线性变换。概率与统计:随机变量、概率分布、分布函数和属性、离散和连续、统计推断 - 估计和假设检验。机器学习(第 1 部分):机器学习基础、线性回归和逻辑回归(分类)。(第 2 部分将在下学期的应用机器学习课程中继续)教材 1. Jiawei Han 和 Micheline Kamber 编写的《数据挖掘概念和技术》 2. Rohatgi 和 Saleh 编写的《概率与统计简介》。 3. Christian Albright 和 Wayne Winston 编写的商业分析:数据分析与决策

蛋白质结构的深层生成模型发现了跨连续折叠空间的远距离关系
我们对折叠空间的看法隐含地取决于许多假设,这些假设影响了我们分析,解释和理解蛋白质结构,功能和进化的方式。例如,查看蛋白质结构的相似性(例如,建筑,拓扑或其他层面)是否有最佳的粒度?同样,折叠空间的离散/连续二分法是中心的,但仍未解决。折叠空间bin“类似”折叠的离散视图分为不同的非重叠组;不可思议,这种融合会错过远程关系。虽然像CATH这样的层次结构系统是必不可少的资源,但较少的启发式和概念上的弹性方法可以实现对折叠空间的更细微的探索。建立在蛋白质结构的“尤其”模型的基础上,在这里,我们提出了一个深层生成建模框架,称为“ deepurfold”,用于分析蛋白质关系。deepurfold的学到的嵌入占据了高维的潜在空间,可以从给定蛋白质上蒸馏而成,以合并的代表统一序列,结构和生物物理特性。这种方法是结构指导的,而不是纯粹基于结构的,而DeepUrfold则学习了代表,从某种意义上说,这些代表“定义”超家族。用CATH部署Deepurfold揭示了逃避现有方法的进化性相关关系,并提出了一种新的,主要是连续的折叠空间视图,这种视图超出了简单的几何相似性,朝着综合序列序列↔结构↔函数↔功能↔函数↔函数↔。