XiaoMi-AI文件搜索系统
World File Search SystemCDK2

197(CDK2 抑制剂)
正在进行的试验:一项首次人体 1a/b 期剂量递增/扩展研究,评估 BG-68501/ETX- 197(CDK2 抑制剂)作为单一疗法或与氟维司群联合治疗 HR+/HER2- 乳腺癌和其他晚期实体瘤患者的效果 作者:Minal Barve、1 Jennifer Man、2 Bruno Fang、3 Alexander Philipovskiy、4 Brian A. Van Tine、5 Rohit Joshi、6 Marion Carrigan、7 Alejandra Ragone、7 Hao Zheng、7 Yang Liu、8 Sally Baron Hay 9 附属机构:1 美国德克萨斯州达拉斯玛丽克劳利癌症研究中心;2 澳大利亚新南威尔士州布莱克敦布莱克敦癌症和血液学中心;3 美国新泽西州东布伦瑞克 Astera 癌症护理中心; 4 佛罗里达癌症专家和研究所/莎拉坎农研究所,美国佛罗里达州玛丽湖;5 华盛顿大学医学院,美国密苏里州圣路易斯;6 南澳大利亚州癌症研究中心,澳大利亚南澳大利亚州阿德莱德;7 百济神州美国公司,美国加利福尼亚州圣马特奥;8 百济神州(上海)有限公司,中国上海;9 悉尼大学,澳大利亚新南威尔士州悉尼 摘要背景:细胞周期蛋白依赖性激酶 (CDK) 2 可通过在 G1/S 和 S/G2 转换期间与细胞周期蛋白 E 或细胞周期蛋白 A 相互作用来调节细胞周期。CDK2 活性升高是 HR+/HER2- 乳腺癌 (BC) 对 CDK4/6 抑制的关键耐药机制。其他基因组改变,例如 RB1 的缺失,可导致其他实体瘤产生耐药性,包括高级别浆液性卵巢癌、胃癌、小细胞肺癌 (SCLC) 和子宫内膜癌。 CCNE1 扩增或细胞周期蛋白 E 过表达可能赋予对 CDK2 抑制的敏感性。BG-68501/ETX-197 是一种强效、选择性的 CDK2 抑制剂,临床前证据表明其在生化和细胞测定中具有强效活性,在癌症异种移植模型中具有显著的抗肿瘤活性,并且对 CDK2 的选择性优于其他 CDK 家族成员。方法:本研究是一项首次在人体中进行的 1a/b 期、开放标签、多中心研究,旨在评估 BG-68501/ETX-197 在晚期、不可切除或转移性实体瘤患者(包括 HR+/HER2- BC)中的安全性、耐受性、PK、药效学和初步抗肿瘤活性。在剂量递增阶段(1a 期),连续队列将接受剂量递增的 BG-68501/ETX- 197 单药治疗或与氟维司群联合治疗;此外,安全性扩展队列将接受推荐剂量的 BG-68501/ETX-197 治疗,以供进一步评估。在剂量扩展阶段(1b 期),HR+/HER2- BC、铂类难治性或耐药性浆液性卵巢癌、输卵管癌、原发性腹膜癌 (PROC)、广泛期 SCLC (ES-SCLC) 或 CCNE1 扩增晚期实体瘤患者将接受 BG-68501/ETX-197 单药口服治疗或与氟维司群联合治疗。资格标准包括年龄≥18 岁、经组织学或细胞学确诊为可能与 CDK2 依赖性相关的晚期或转移性实体瘤的患者,这些患者已接受过≥1 线局部晚期或转移性疾病治疗,并且之前接受过内分泌治疗,并且在辅助治疗或局部晚期或转移性环境中接受过 CDK4/6 抑制剂治疗 HR+/HER2- BC,或之前接受过所有其他晚期实体瘤的标准治疗。对于剂量递增阶段(第 1a 阶段),主要目标是评估 BG-68501/ETX- 197 单药治疗或与氟维司群联合治疗的安全性和耐受性,并确定最大耐受剂量、最大给药剂量和推荐扩增剂量 (RDFE);次要目标是评估研究者按照 RECIST v1.1 评估的初步抗肿瘤活性(ORR、缓解持续时间 [DoR]、至缓解时间 [TTR]、疾病控制率 [DCR] 和临床受益率 [CBR])和 PK。对于剂量扩展阶段(1b 期),主要目标是评估 BG-68501/ETX-197 与氟维司群联合治疗 HR+/HER2- 晚期或转移性 BC 患者的抗肿瘤活性 (ORR),以及 BG-68501/ETX-197 单药治疗 PROC、ES-SCLC 和其他伴有 CCNE1 扩增的晚期或转移性实体瘤患者的抗肿瘤活性 (ORR);次要目标是进一步评估 BG-68501/ETX- 197 单独用于治疗前述晚期实体瘤或与氟维司群联合用于治疗
inx-315,一种有效的选择性CDK2抑制剂,证明了强大的
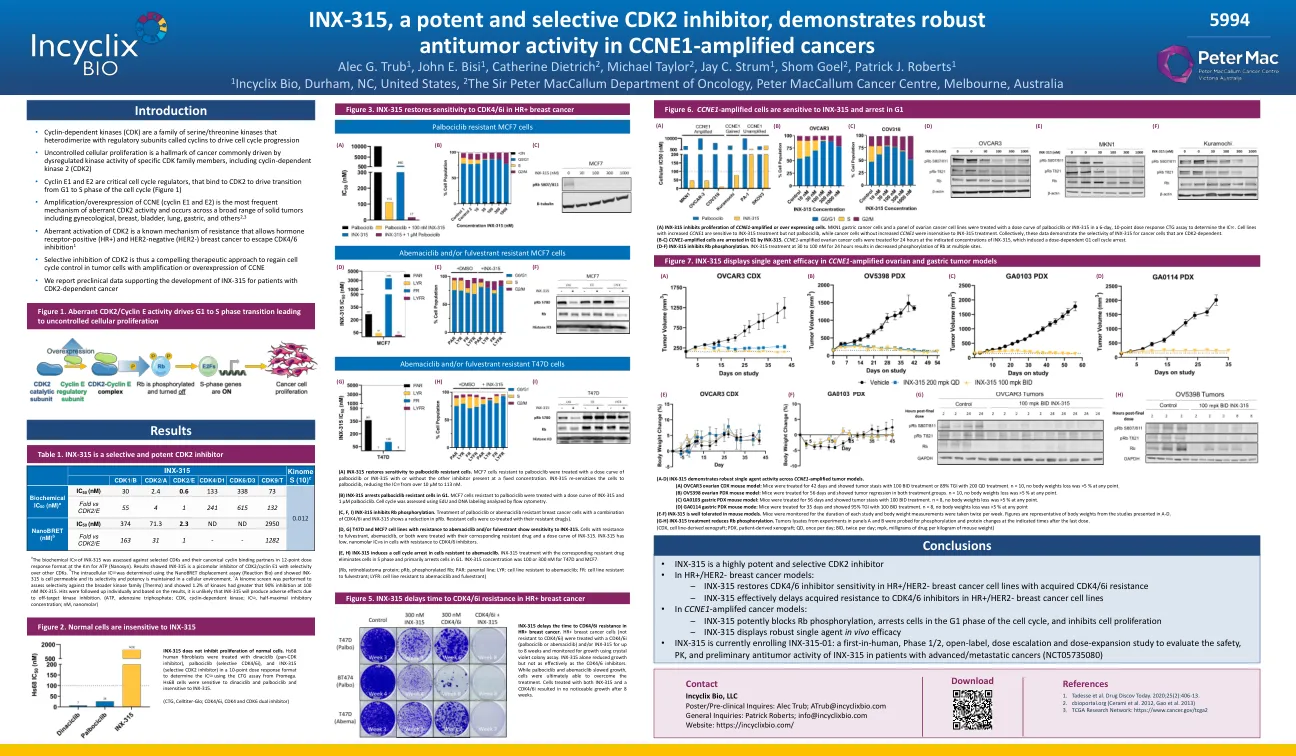
(a)INX -315抑制CCNE1扩增或超出表达细胞的增殖。MKN1胃癌细胞和一组卵巢癌细胞系在6天的10点剂量反应CTG分析中用palbociclib或INX-315的剂量曲线处理,以确定IC 50。CCNE1增加的细胞系对INX-315治疗敏感,而不是palbociclib,而没有增加CCNE1的癌细胞对INX-315治疗不敏感。 共同证明了INX-315对CDK2依赖性的癌细胞的选择性。 (B-C)CCNE1扩增细胞在G1中通过INX-315停止。 CCNE1-放大卵巢癌细胞在指定的INX-315浓度下处理24小时,这诱导剂量依赖性G1细胞周期停滞。 (D-F)INX-315抑制RB磷酸化。 INX-315在30至100 nm的处理24小时下处理导致多个位点的RB磷酸化降低。CCNE1增加的细胞系对INX-315治疗敏感,而不是palbociclib,而没有增加CCNE1的癌细胞对INX-315治疗不敏感。共同证明了INX-315对CDK2依赖性的癌细胞的选择性。(B-C)CCNE1扩增细胞在G1中通过INX-315停止。 CCNE1-放大卵巢癌细胞在指定的INX-315浓度下处理24小时,这诱导剂量依赖性G1细胞周期停滞。 (D-F)INX-315抑制RB磷酸化。 INX-315在30至100 nm的处理24小时下处理导致多个位点的RB磷酸化降低。(B-C)CCNE1扩增细胞在G1中通过INX-315停止。CCNE1-放大卵巢癌细胞在指定的INX-315浓度下处理24小时,这诱导剂量依赖性G1细胞周期停滞。(D-F)INX-315抑制RB磷酸化。INX-315在30至100 nm的处理24小时下处理导致多个位点的RB磷酸化降低。INX-315在30至100 nm的处理24小时下处理导致多个位点的RB磷酸化降低。
BLU-222 是一种口服、强效且选择性的 CDK2 抑制剂,...
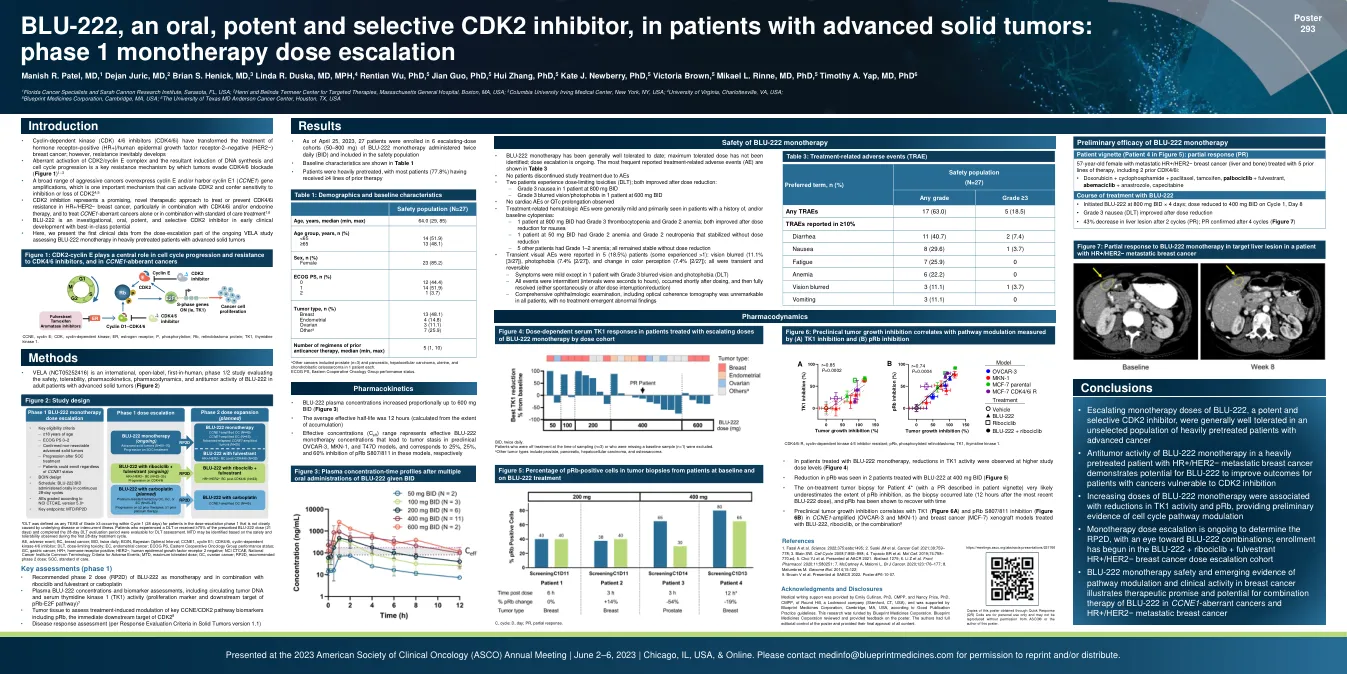
• 细胞周期蛋白依赖性激酶 (CDK) 4/6 抑制剂 (CDK4/6i) 改变了激素受体阳性 (HR+)/人类表皮生长因子受体 2 阴性 (HER2-) 乳腺癌的治疗;然而,耐药性不可避免地会产生 • CDK2/细胞周期蛋白 E 复合物的异常激活以及由此导致的 DNA 合成和细胞周期进展的诱导是肿瘤逃避 CDK4/6 阻断的关键耐药机制(图 1)1-3 • 多种侵袭性癌症过度表达细胞周期蛋白 E 和/或携带细胞周期蛋白 E1(CCNE1)基因扩增,这是一种可激活 CDK2 并赋予对 CDK2 抑制或缺失的敏感性的重要机制 4,5 • CDK2 抑制代表了一种有前途的新型治疗方法,可用于治疗或预防 HR+/HER2-乳腺癌中的 CDK4/6i 耐药性,特别是与 CDK4/6i 和/或内分泌疗法相结合,以及单独或与标准治疗方法相结合治疗 CCNE1 异常癌症 1,6 • BLU-222 是一种在研的、口服的、强效的、选择性的 CDK2 抑制剂,处于早期临床开发阶段,具有同类最佳的潜力 • 在这里,我们介绍了第一个正在进行的 VELA 研究剂量递增部分的临床数据,该研究评估了 BLU-222 单药治疗接受过大量治疗的晚期实体瘤患者

癌症中 CDK2 靶向治疗的挑战和机遇
细胞周期依赖性激酶 2 (CDK2) 在细胞周期调控中起着关键作用,并参与一系列生物过程。CDK2 与 DNA 损伤、细胞内运输、蛋白质降解、信号转导、DNA 和 RNA 代谢和翻译等途径中的蛋白质相互作用并对其进行磷酸化。CDK2 及其调节亚基在许多人类癌症中失调,有新证据表明 CDK2 抑制会在具有明确遗传特征的一组肿瘤中引发抗肿瘤活性。以前的 CDK2 抑制剂是非特异性的,并且受到脱靶效应的限制。新一代 CDK2 抑制剂的开发为 CDK2 依赖性癌症的治疗提供了机会。

CDK2 抑制剂作为抗癌药物的最新进展
细胞周期依赖性激酶 (CDK) 是一组丝氨酸/苏氨酸激酶,它们与称为细胞周期蛋白的调节亚基相互作用以发挥其活性。1,2 这些 CDK 在调节细胞周期进程和转录方面至关重要(►图 1)。3,4 在人类细胞中已鉴定出 20 种 CDK(CDK1 – 20)和 29 种细胞周期蛋白。5 其中,CDK1、CDK2、CDK4 和 CDK6 在细胞周期调控中起关键作用,而 CDK7 – 9、11 – 13、19 和 20 主要在转录调控中起关键作用。2,4,6,7 值得注意的是,许多 CDK 具有多种催化底物并参与各种细胞过程。例如,CDK7 是一种在细胞周期期间激活 CDK 的激酶,也是转录期间 RNA 聚合酶 II 的调节剂。 CDK5 被广泛认为是调节神经元功能和细胞迁移的关键因素。8 癌症的一个标志性特征是细胞周期失调,导致不受控制和过度的细胞

Cyclin E1/CDK2激活是妇科癌症中WEE1激酶抑制的关键弱点
附加声明:存在利益冲突 D. Kim 是 Zentalis Pharmaceuticals 的员工和股东。H. Chung 是 Zentalis Pharmaceuticals 的员工和股东。W. Liu 是 Zentalis Pharmaceuticals 的员工和股东。S. Kim 是 Zentalis Pharmaceuticals 的员工和股东。X. Guo 是 Zentalis Pharmaceuticals 的员工和股东。N. Jameson 是 Zentalis Pharmaceuticals 的员工和股东。P.R.de Jong 是 Zentalis Pharmaceuticals 的前员工。S. Yea 是 Zentalis Pharmaceuticals 的员工和股东。L. Harford 是 Zentalis Pharmaceuticals 的员工和股东。J. Li 是 Zentalis Pharmaceuticals 的前雇员。D. Kim 是 Zentalis Pharmaceuticals 的员工和股东。K. Fischer 是 Zentalis Pharmaceuticals 的员工和股东。A. Samatar 是 Zentalis Pharmaceuticals 的员工,也是 Zentalis Pharmaceuticals 的股东。A. Jubb 是 Zentalis Pharmaceuticals 的员工和股东。K. Bunker 是 Zentalis Pharmaceuticals 的员工,也是 Zentalis Pharmaceuticals 的股东。K. Blackwell 是 Zentalis Pharmaceuticals 的员工和股东。F. Simpkins 是阿斯利康、葛兰素史克和 Zentalis Pharmaceuticals 的科学顾问委员会成员;已获得阿斯利康、Repare Therapeutics、Instill Bio 和 Sierra Oncology 的机构研究资助。F. Meric-Bernstam 是以下公司的顾问。G.B.Mills 是 Amphista、Astex、阿斯利康、BlueDot、Chrysallis Biotechnology、Ellipses Pharma、GSK、ImmunoMET、In¬nity、Ionis、Leapfrog Bio、Lilly、Medacorp、Nanostring、Nuvectis、PDX Pharmaceuticals、Qureator、Roche、Signalchem Lifesciences、Tarveda、Turbine、Zentalis Pharmaceuticals 的科学顾问委员会/顾问;股票/期权/财务:Bluedot、Catena Pharmaceuticals、ImmunoMet、Nuvectis、SignalChem、Tarveda、Turbine;许可技术:Myriad Genetics 的 HRD 检测、Nanostring 的 DSP 专利;赞助研究:阿斯利康。O. Harismendy 是 Zentalis Pharmaceuticals 的员工和股东。J. Ma 是 Zentalis Pharmaceuticals 的员工和股东。M.R.Lackner 是 Zentalis Pharmaceuticals 的员工和股东。其他作者未报告任何披露。AbbVie、Aduro BioTech Inc.、Alkermes、阿斯利康、第一三共株式会社、Calibr(斯克里普斯研究公司旗下的一个部门)、DebioPharm、Ecor1 Capital、eFFECTOR Therapeutics、Exelixis、F. Hoffman-La Roche Ltd.、GT Apeiron、Genentech Inc.、Harbinger Health、IBM Watson、Incyte、In¬nity Pharmaceuticals、Jackson Laboratory、Jazz Pharmaceuticals、Kolon Life Science、LegoChem Bio、Lengo Therapeutics、Loxo Oncology、Menarini Group、OrigiMed、PACT Pharma、Parexel International、P¬zer Inc.、Protai Bio Ltd、Samsung Bioepis、Seattle Genetics Inc.、Tallac Therapeutics、Tyra Biosciences、Xencor、Zymeworks; Black Diamond、Biovica、Eisai、FogPharma、Immunomedics、Inection Biosciences、Karyopharm Therapeutics、Loxo Oncology、Mersana Therapeutics、OnCusp Therapeutics、Puma Biotechnology Inc.、Seattle Genetics、Sano¬、Silverback Therapeutics、Spectrum Pharmaceuticals、Theratechnologies、Zentalis 的咨询委员会;获得 Jazz Pharmaceuticals、Zymeworks、Aileron Therapeutics, Inc. AstraZeneca、Bayer Healthcare Pharmaceutical、Calithera Biosciences Inc.、Curis Inc.、CytomX Therapeutics Inc.、Daiichi Sankyo Co. Ltd.、Debiopharm International、eFFECTOR Therapeutics、Genentech Inc.、Guardant Health Inc.、Klus Pharma、Takeda Pharmaceutical、Novartis、Puma Biotechnology Inc.、Taiho Pharmaceutical Co. 的赞助研究(对机构而言);Dava Oncology 的酬金;获得欧洲癌症研究与治疗组织 (EORTC)、欧洲肿瘤医学学会 (ESMO)、胆管癌基金会、Dava Oncology 的旅行相关资金和报销。

INCB123667(一种选择性CDK2抑制剂)的安全性和耐受性,对实体瘤的患者:1期研究
注意:C1D8 PK数据是从0-8 h收集的,从0-8 h,QD收集0-24 h。在12小时的时间点再次使用了出价的预剂量样品。对于出价,将第一个剂量后的数据绘制在0-12 h之间,然后从12-24小时再次绘制,以在第二剂剂量后复制轮廓。50 mg QD(n = 5); 50 mg竞标(n = 19); 75 mg QD(n = 19); 125 mg QD(n = 20); 150 mg QD(n = 5)。平均值±SD CDK2 IC 50 = 570±468(n = 66);平均值±SD CDK1 IC 50 = 5488±1759(n = 6)(全血分析)。*由PredicinesCore估算的副本编号

开发CDK2选择性的小分子抑制剂...
Primary resistance: About 15% of patients treated with CDK4/6 inhibitor (CDK4/6i) + aromatase inhibitor, and up to 30% of those treated with CDK4/6i + fulvestrant, will develop recurrent disease within 6 months Acquired resistance: Almost all patients will eventually develop progressive disease Multiple pathways are implicated in resistance CCNE1 amplification and cyclin E1 overexpression are
Blu-222是一种有效且高度选择性的CDK2抑制剂,在CCNE1-异常中表现为抗肿瘤活性,作为单一疗法和组合治疗
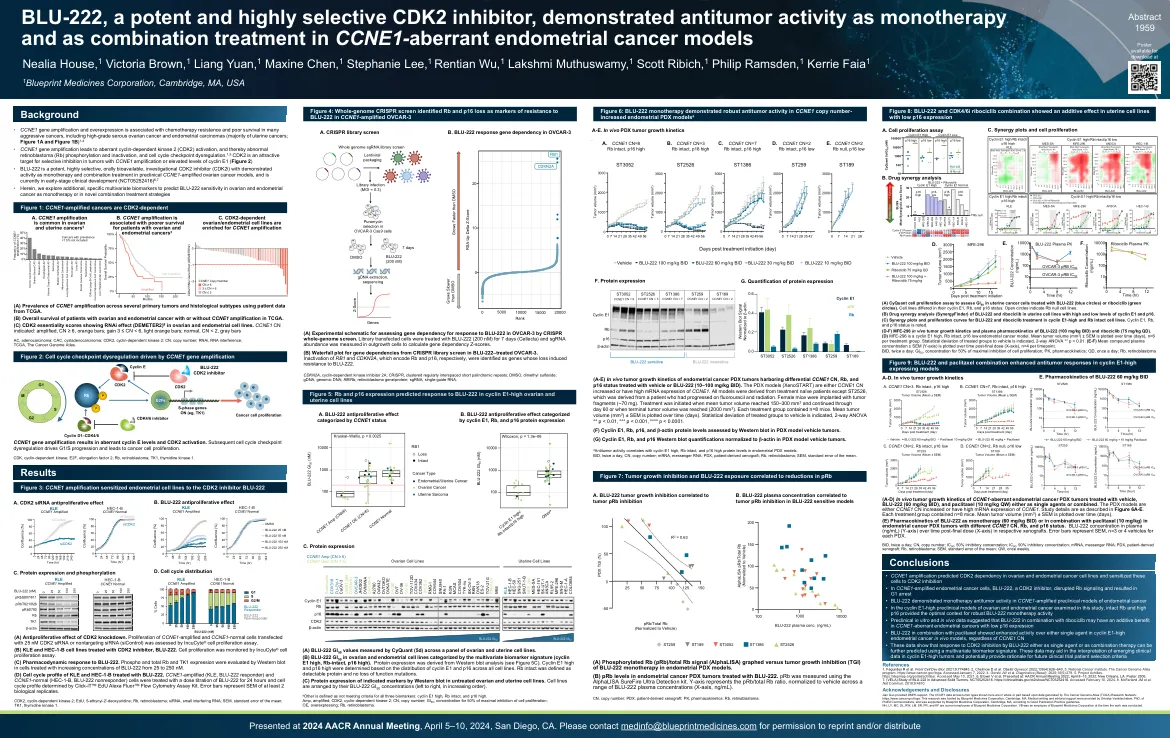
(A-E)子宫内膜癌的体内肿瘤生长动力学PDX肿瘤具有差异CCNE1 CN,RB和P16状态,该状态已使用媒介物或BLU-222处理(10–100 mg/kg BID)。PDX模型(Xenostart)是CCNE1 CN增加或具有CCNE1的mRNA表达高。所有模型均来自治疗幼稚的患者,除了ST2526,该患者源自氟尿嘧啶和放射线的患者。雌性小鼠植入肿瘤碎片(〜70 mg)。当平均肿瘤体积达到150-300 mm 3并持续到第60天或达到末端肿瘤体积时,开始治疗(2000毫米3)。每个治疗组均包含n = 8只小鼠。平均肿瘤体积(mm 3)±SEM随时间(天)绘制。指示了处理的组向车辆的统计偏差,2路ANOVA ** P <0.01,*** p <0.001,**** p <0.0001。

PF-07104091,一种级别的CDK2-选择性抑制剂...
通过选择性CDK4/6抑制剂在HR+/HER2-乳腺癌中选择性抑制细胞周期进展的选择性抑制作用。通过选择性抑制CDK2扩大对细胞周期的控制提供了癌症中新型的治疗机会,包括靶向CCNE1扩增肿瘤和对ER+乳腺癌中CDK4 \ 6抑制剂的抵抗力。PF-07104091是临床研究,是临床研究的第一类CDK2选择抑制剂。使用PF-07104091进行临床前研究,确定了CDK2抑制在疾病中的治疗影响,并突出了CDK2在控制癌细胞增殖中的不同机械作用。 在CCNE1扩增的卵巢癌模型中,CDK2在控制RB1磷酸化和G1检查点中起主要作用。 用PF-07104091抑制CDK2诱导G1生长停滞,并控制肿瘤异种移植作为单药治疗。 在ER+乳腺癌模型中,CDK2在控制RB1磷酸化的控制中起支持作用,并与CDK4 \ 6合作。 与CDK4 \ 6抑制作用结合使用CDK2 KO作为CDK4 \ 6抑制作用的主要敏化剂,并支持Cyclin E \ CDK2复合物作为对CDK4 \ 6抑制剂的抗CDK4 \ 6抑制剂的驱动力,整个基因组CRIS敲除(KO)和CRISPR激活筛选与CDK4 \ 6抑制作用建立了CDK2 KO作为抑制CDK4 \ 6抑制作用。使用PF-07104091进行临床前研究,确定了CDK2抑制在疾病中的治疗影响,并突出了CDK2在控制癌细胞增殖中的不同机械作用。在CCNE1扩增的卵巢癌模型中,CDK2在控制RB1磷酸化和G1检查点中起主要作用。用PF-07104091抑制CDK2诱导G1生长停滞,并控制肿瘤异种移植作为单药治疗。 在ER+乳腺癌模型中,CDK2在控制RB1磷酸化的控制中起支持作用,并与CDK4 \ 6合作。 与CDK4 \ 6抑制作用结合使用CDK2 KO作为CDK4 \ 6抑制作用的主要敏化剂,并支持Cyclin E \ CDK2复合物作为对CDK4 \ 6抑制剂的抗CDK4 \ 6抑制剂的驱动力,整个基因组CRIS敲除(KO)和CRISPR激活筛选与CDK4 \ 6抑制作用建立了CDK2 KO作为抑制CDK4 \ 6抑制作用。用PF-07104091抑制CDK2诱导G1生长停滞,并控制肿瘤异种移植作为单药治疗。在ER+乳腺癌模型中,CDK2在控制RB1磷酸化的控制中起支持作用,并与CDK4 \ 6合作。整个基因组CRIS敲除(KO)和CRISPR激活筛选与CDK4 \ 6抑制作用建立了CDK2 KO作为抑制CDK4 \ 6抑制作用。PF-07104091 combined with CDK4\6 inhibitor palbociclib or CDK4-selective inhibitor PF-07220060 synergistically controls proliferation of ER+ BC cells in vitro and induces tumor regression in ER+ BC xenograft models, including PDX models with acquired resistance to CDK4\6 inhibitors and endocrine therapy.