XiaoMi-AI文件搜索系统
World File Search SystemCGTP

附录 4 - 生物制品注册指南。......
细胞和基因治疗产品 (CGTP) 属于生物制品监管范畴。与主要为纯化细胞蛋白的生物技术产品不同,CGTP 含有活细胞和功能性细胞。因此,CGTP 受单独框架监管。详情请参阅马来西亚细胞和基因治疗 (CGTP) 注册指导文件和指南。本文件为制造商、申请人、医疗保健专业人员和公众提供有关马来西亚 CGTP 注册法律安排的信息。根据 2017 年第 6 号指令,该指南将于 2021 年 1 月 1 日强制实施。有关分阶段注册和执行 CGTP 的机制详情,请参阅 2020 年第 19 号指令。参考资料: i) 第 19 号指令,2020. NPRA.600-1/9/13(10) Direktif Berkenaan Pelaksanaan Pendaftaran Produk dan Penguatkuasaan Secara Berperingkat Bagi Produk Terapi Sel dan Gen (CGTPs) Serta Tambahan Senarai Produk Di Luar Skop Kawalan CGTPs Oleh PBKD(2020 年 12 月 14 日)

药品制造中的人工智能 - OPQ_人工智能讨论档案编号 FDA-2023-N-0487 - 反馈
1271.160(d):“…如果您依赖软件来遵守核心 CGTP 要求,并且软件是定制软件或已定制或编程(包括编程为执行用户定义的计算或表格的软件)以执行与核心 CGTP 要求相关的功能的商业可用软件,则您必须验证计算机软件的性能是否符合预期用途,以及对该软件进行的任何更改是否符合预期用途。您必须验证所有其他软件的性能是否符合预期用途”
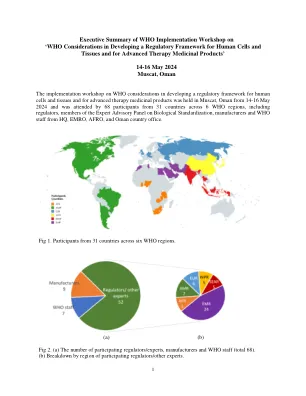
执行谁实施研讨会
在为期三天的研讨会中,在最近发表的文档中概述了“在为人类细胞和组织开发监管框架以及晚期治疗药物产品的考虑方面的考虑因素”,并在七个公开会议中进行了讨论。研讨会的议程已包含在本文档的末尾。细胞,组织和基因治疗产物(CGTP)代表了对复杂性的生物产物,来自那些需要最小的监督(例如简单的同种异体皮肤移植物)(例如嵌合抗原受体T细胞)的生物学产品,这些抗原受体T细胞需要大量的调节性评估以证明产品安全性和效率在营销授权之前。在研讨会上讨论了生物产品的光谱和关键风险考虑因素。讨论了一种基于风险的CGTP,分类,CGTP的术语以及全球协调的重要性,尤其是在术语上的重要性。会议包括讨论CGTP的监管方法,分类和术语的考虑,以及国家监管局(NRA)代表对国家监管景观的观点。制造商对当前CGTP开发状态的看法也在会议上进行了讨论,以及谁在支持国家加强其监管系统并建立技术能力以调节CGTP的国家中的作用。讲习班总结了建议的下一步。表1。基于风险的监管方法的CGTP案例研究小组活动包括对三个案例研究的讨论:1)最低生产的组织,用于接受者与供体相同功能:受辐射的,无菌的人类皮肤皮肤,2)需要全面的预上市授权的生物产品,并证明安全性和有效性:自体CAR T细胞产品靶向CD19和3)在受益方面和3)与受体相同的plotic and Is functore n和Donor的pant portor:Allog:Allog and donor:Allog:Allog:Allog:Allog nog poror:Allog and prore portor:Allog and prore portor:Allog and donor portor:间充质干细胞(MSC)。总体而言,对基于案例的研究的审查重申了对基于风险的细胞和基因治疗产品分类方法的紧迫需求,并且需要提高制造依从性(GTP vs GMP)使用较高风险产品的需求。参与者强调,迫切需要对最小操纵和同源用途进行协调的定义,以防止误解,因为对监管框架的不同解释(或缺乏监管框架)的不同解释可能会使“不良玩家”对“不良玩家”的吸引力,以使其对细胞和基因治疗市场更具吸引力,并且可能对患者产生损害。还提出了至关重要的一点,即需要与教育和提供足够的信息同时完成此类监管框架。

干细胞疗法产品和再生医学的标准
3.1.1.1由DOH许可为医疗机构。 只有DOH许可的医疗机构才有资格提供干细胞,细胞和基于组织的疗法。 3.1.1.2拥有一个机构干细胞研究委员会(ISCRC)或当地研究伦理委员会(REC),能够提供与治疗和/或产品有关的专门内部监督机制。 本地ISCRC/REC将是寻求ADHRTC批准的唯一负责机构。 3.1.1.3该疗法得到信誉良好的干细胞组织的批准,HCF在已知的国际干细胞研究实体之一中具有国际认证/注册。 3.1.1.4确保DOH许可设施或GMP认证的设施执行干细胞和衍生工具的采购(收集,生物库访问或导入)。 3.1.1.5确保通过DOH许可的实验室来处理干细胞和衍生物的处理,用于制造工艺,这可以是现场或现场。 3.1.1.6遵守现有的法规和准则,以确保产品安全,纯净且有效地满足CGTP,GMP和GCP要求。 3.1.1.7在应用干细胞,细胞和基于组织的疗法之前,获得ADHRTC和ISCRC或当地研究伦理委员会(REC)的批准。3.1.1.1由DOH许可为医疗机构。只有DOH许可的医疗机构才有资格提供干细胞,细胞和基于组织的疗法。3.1.1.2拥有一个机构干细胞研究委员会(ISCRC)或当地研究伦理委员会(REC),能够提供与治疗和/或产品有关的专门内部监督机制。本地ISCRC/REC将是寻求ADHRTC批准的唯一负责机构。3.1.1.3该疗法得到信誉良好的干细胞组织的批准,HCF在已知的国际干细胞研究实体之一中具有国际认证/注册。3.1.1.4确保DOH许可设施或GMP认证的设施执行干细胞和衍生工具的采购(收集,生物库访问或导入)。3.1.1.5确保通过DOH许可的实验室来处理干细胞和衍生物的处理,用于制造工艺,这可以是现场或现场。3.1.1.6遵守现有的法规和准则,以确保产品安全,纯净且有效地满足CGTP,GMP和GCP要求。3.1.1.7在应用干细胞,细胞和基于组织的疗法之前,获得ADHRTC和ISCRC或当地研究伦理委员会(REC)的批准。

处方药用户费用法(PDUFA)重新授权
目的是讨论FDA和行业CBER特定的增强建议。参与者FDA行业Rachael Anatol Cber E. Cartier E. Cartier Bio Angela Granum Cber Brad Glasscock(Lead)Bio(Biomarin)Chris Joneckis(FDA Lead)Cber Mathias Mathias Hukkelhoven Phrma(BMS) Bio(Gilead and Kite)Darlene Martin Cber Lucy Vereshchagina Phrma Carol rehkopf Cber PDUFA VII CBER分组分组讨论集中在CBER中的细胞和基因治疗(CGT)计划。CBER细胞和基因治疗计划资源分配CBER提供了有关细胞和基因治疗计划(CGTP)所需位置的其他信息,包括支持,间接和直接审查角色的位置。cber指出,指导开发是资源请求中包括的间接成本的一部分。cber定期发布指导列表,以指示给定年份出版的预期。cber将讨论本提案要求的登机位置的分配和排序。cber提供了有关当前职位的数据,并讨论了平衡资源的方法,例如实现主管与员工的最佳比例。还讨论了新资源的招聘,招聘和培训,并且是CBER的优先事项。FDA提出,雇用的目标是雇用新资源并尽快雇用船上的目标。在登机后,新的审阅者需要培训并经验丰富的熟练程度。CBER将为现有和新员工的培训需求提供估算。其他FDA估计值,包括为2023财年提供预测提交的类型和数量。cber解释说,预测是保守的,预测模型中使用的2020财年数据将被更新。CBER还将刷新建模数据。

便捷注册途径指南
1.1 背景 有效的监管体系是加强卫生系统的重要组成部分,有助于改善公共卫生结果。为了提高监管能力和效率,国家药品监管机构 (NPRA) 于 2019 年发布了第一版简化注册途径 (FRP) 指南。该指南基于依赖概念制定。依赖机制的应用充分利用了其他监管机构对旨在投放当地市场的相同产品所做的工作。启动该机制是为了减少重复工作,并使 NPRA 能够在上市前和上市后授权活动中强调基于风险的方法。依赖程序提供了更高效的审查流程,并最终使药品能够尽早进入市场。 1.2 目标 本指导文件旨在提供一个透明且一致的依赖方法利用程序。该指南描述了通过该途径提交产品注册申请的程序和要求,并作为药品注册指南文件 (DRGD) 的补充文件。申请人应在通过 QUEST 系统完成产品注册申请之前参考这两份文件。1.3 范围本指南的范围适用于新药产品、仿制药和生物制剂,包括细胞和基因治疗产品 (CGTP)。最终是否通过 FRP 考虑产品取决于 NPRA 的判断。NPRA 可根据本指南中规定的资格标准做出决定,其中可能包括风险收益评估、国家优先事项、公共卫生需求以及其他依赖原因或机会。

基因编辑产品效力测定
CASSS 细胞和基因治疗产品 (CGTP) 2024 研讨会是一年一度的活动,行业、监管和学术专业人士将在会上讨论与细胞和基因治疗领域相关的挑战和最新动态。会议举办了各种全体会议和圆桌会议,以鼓励讨论从可比性和制造到基因组编辑技术和 ICH 指南等各种主题。一场受欢迎的全体会议是关于基因编辑产品的效力测定。该会议包含 3 个演讲,随后是小组讨论,由观众提问。第一位演讲者是 FDA 生物制品评估和研究中心 (CBER) 的 Andrew Byrnes 博士。他的演讲介绍了有关细胞和基因治疗产品效力保证的新指导文件草案的信息。他强调,效力保证策略 (PAS) 不仅仅涉及效力测定,而且成功的策略应该涵盖效力的所有方面。因为 PAS 是“一种综合方法,有助于确保每一批产品都具有实现预期治疗效果所需的效力”,1 Byrnes 博士详细说明,PAS 需要申办方了解并对其产品特有的效力相关特性进行风险评估,降低效力相关关键质量属性的风险,并在对产品和制造工艺有更多了解后重新评估和改进 PAS。本指南草案包括与 PAS 相关的所有方面的建议和一般建议,包括效力测定,特别是关于其使用、开发和验收标准。继 Byrnes 博士之后,ElevateBio 的 Debaditya Bhattacharya 博士介绍了针对亨廷顿氏病 (HD) 的体内 AAV 基因编辑疗法的效力测定的开发。作为 ElevateBio 的分析开发副总裁,他负责各种不同模式的 CMC 分析开发、策略和测试操作。考虑到试验性亨廷顿氏病疗法的预期活动,Bhattacharya 博士讨论了效力测定的开发策略以及如何确定属性(例如确定合适的细胞系)。在这种情况下考虑的主要属性是 1) 它是否包含用于 RNP 靶标接合的目标 SNP,2) 其 AAV 转导效率,以及 3) 在培养中生长和维持的“容易程度”。通过进行桑格测序以筛选 SNP、流式细胞术以访问转导以及