XiaoMi-AI文件搜索系统
World File Search SystemCHMP

Braftovi,Inn -Corafenib-欧洲药品局
1。EMA/CVMP。2022:2。欧洲指令2001/82/EC指令2001/82/EC的2019/6。2019:https:// eur-lex。3。EMA/CVMP。2021:/www.europa.eu.eu/en/cvmp-strategy-anticrobial-2025_en.pdf4。EMA和HMA,欧洲医学AGWork战略策略至2025年。公共保护变更(EMA/8501/2020)。2020:https://www.europ.eu/european/european-public-free-public-public-public-merican。5。EMA,2025(EMA/110706/2020)。2020:https://www.europa.eu/en/documents/reflection-procedury-2025-strategic-reflection_en.pdf6。EMA/CVMP。2021:/www.europa.eu.eu.eu/en-producted-spc-spec-products特定特定特定的特定特定特定产品7。欧洲药品局和抗菌建议临时专家小组(AMEG),欧盟抗生素的分类(EMA/CVMP/CVMP/CHMP/682198/2017)。2019:https://www.ema.europa.eu/en/documents/report/categorisation-categorisation-antibiotics-europearics-european-union- onsect-request-request-european-commission-commission-commission-commission-commission-complatific_en.pdf。

报告-Bioarctic
2024年第四季度的活动•澳大利亚药品局(TGA)决定不批准李卡纳姆布。Eisai has requested a reconsideration • The phase 3 study AHEAD 3-45 in preclinical Alzheimer's disease was fully recruited • Eisai completed the stepwise application for subcutaneous maintenance therapy with Leqembi in the US • New data for the BrainTransporter technology was presented, showing a dramatic increase of antibody delivery to the brain • Eisai lowered Leqembi outlook for fiscal year 2024年(2024年4月 - 2025年3月)。销售预计将达到JPY 42.5 B•EMA的咨询委员会CHMP发出了积极的建议,以批准欧盟的LeCanemab•Bioarctic与Bristol Myers Squibb(BMS)签署了一项全球独家许可协议(BMS),以提供生物极低的抗体的BAN1503和BAN2803。该协议正在等待批准,值得超过1.35 B加版本

工作计划非临床域2025-2027_final
1S1a一种物质一评估3RS替换,减少和改进3RSWP替代,减少和精致工作组AI人工智能ATMP ATMPS高级治疗药物BRT OEG批处理释放oeg批次释放运营专家小组CAT委员兽医药物相互认可和分散程序的协调组CPCA致癌性效力分类方法CTCG临床试验临床试验兽医药物dili药物诱导肝损伤的临床试验小组CVMP委员会CVMP委员会

新闻稿 Sarclisa 在欧盟获批,成为首个与标准治疗 VRd 联合用于治疗不适合移植的淋巴细胞白血病的抗 CD38 疗法
• 该批准基于 IMROZ 3 期研究的积极结果,该结果证明 Sarclisa 与标准治疗方法相结合可显着改善 TI NDMM 的 PFS,而单独使用标准治疗方法则不然 • 代表欧盟的第三种适应症,其中两种用于治疗 R/R MM 成年患者,一种用于治疗 NDMM 巴黎,2025 年 1 月 22 日。在欧洲药品管理局 (EMA) 人用药品委员会 (CHMP) 采取积极意见后,欧盟已批准 Sarclisa 与标准治疗方法硼替佐米、来那度胺和地塞米松 (VRd) 相结合,用于治疗不适合自体干细胞移植 (ASCT) 的新诊断多发性骨髓瘤 (NDMM) 成年患者,基于来自 IMROZ 3 期研究的数据。随着扩大营销授权,Sarclisa 成为欧盟首个针对该患者群体与 VRd 联合使用的抗 CD38 疗法。

IPSC衍生的视网膜色素上皮...
1美国FDA,2023年8月。建议可接受的亚硝基药物药物相关杂质的摄入量限制(NDSRIS)https://www.fda.gov/media/170794/下载2欧洲药品局(EMA),2024年1月。对营销授权持有人/申请人的提问和答案是关于人类药用产品中硝基胺杂质的第726/2004条第5条第3款(EC)第5条第3款的意见。EMA/409815/2020 REV20。https://www.ema.europa.eu/en/documents/referral/referral/nitrosamines-emea-h-a53-1490-questions-questions-questions-questions-markwers-marketing-marketing--authorisation-markaret-holders--holders-holders-holders-holders-holders-holders-holders-holders-holders-holders--plicant-chmp-plicant-chmp-chmpopopiminiion-条款-53-ECEC-NO-726/2004-硝基胺 - 含量 - 含量 - 烟 - 中间人 - products_en.en.pdf 3欧洲药品局(EMA),2019年,2019年。评估报告:根据指令2001/83/EC的第31条的转诊:包含四唑组的血管紧张素-II受体拮抗剂(Sartans)。EMA/217823/2019。https://www.ema.europa.eu/en/documents/variation-report-report/angiotensin-ii-ii-ii-receptor-antagonists-sartans-sartans-article-31- referral-crefral-chmp-chmp-shmp-sassessment-report_en.pdf 4 swissmedic,2019年。潜在的亚硝基胺污染:请求进行风险评估。https://www.swissmedic.ch/swissmedic/en/home/news/mitteilungen/aufforderung-zlinhaberinnen-ham.htm l。

BW Energy -2024年度报告
积极的CHMP意见基于3A阶段临床试验计划的结果。与2型糖尿病患者的每日基底胰岛素相比,每周的基础胰岛素ICODEC降低了较高的血糖降低1(通过HBA 1C的变化来衡量)和较好的时间(在推荐的血糖范围内花费的时间)。在以前尚未接受胰岛素治疗的2型糖尿病患者中,总体观察到的临床意义或严重低血糖3的速率低于每个患者年度暴露年度的一个事件,均为每周一次的基础胰岛素ICODEC和比较剂和比较剂。在患有1型糖尿病的患者中,曾经每周的基础胰岛素ICODEC在降低HBA 1C时表现出非劣质性,与胰岛素degludec 4相比,严重或临床意义高度低血糖的统计学意义上的较高估计率更高。在整个程序中,每周一次的基础胰岛素ICODEC似乎具有安全且耐受性良好的轮廓。

MR_CHMP建议欧盟批准Phesgo_en
•phesgo在短短几分钟内,在皮肤下提供更快,更少的侵入性提供护理标准治疗,而静脉输注1,2,3•皮下给药的偏爱是患者,医生和医疗保健提供者的首选,并且可以与医院的时间和成本相关联,这是•第一个概述的人群•同时是•近期•同时,这是两次comploy•hiptib y him in tros in comploy intib•himbon tros in comploy intib y 5,5,5,6• 2020年11月13日,由单一皮下注射巴塞尔(Roche)(六:RO,ROG; OTCQX:ROG; OTCQX:RHHBY)今天宣布,欧洲药品局(EMA)人类使用药物委员会(CHMP)推荐了Perjeta®的固定剂量组合(perjetaiumabimabimabimabimabimabimabimabimab)透明质酸酶,由皮下(SC;在皮肤下)和静脉内(IV)化学疗法结合使用,以治疗早期和转移性HER2阳性乳腺癌。基于这一建议,预计欧盟委员会在不久的将来有望获得有关Phesgo的最终决定。“我们致力于改变乳腺癌患者生活的承诺远远超出了改善疗效结果,”罗氏首席医学官兼全球产品开发负责人Levi Garraway博士说。“今天的建议是重新定义欧洲患有HER2阳性乳腺癌患者的护理标准的又一重要一步,它可能会提供一种更快,更侵入性的方式来接受Perjeta和Herceptin接受治疗。” Phesgo的SC给药大约需要八分钟的初始加载剂量,每个随后的维持剂量大约需要五分钟。1与使用标准IV配方输注perjeta和Herceptin的载荷剂量的大约150分钟相比,随后对两种药物进行维护输注60-150分钟。2,3 CHMP的建议是基于III期Federica研究的结果,该研究表明,与两种药物相比,使用Phesgo的治疗在血液中产生了非内部perjeta和Herceptin。对化学疗法的Phesgo的安全性与Perjeta Plus Herceptin和化学疗法的静脉注射疗法相当,并且没有发现新的安全信号,包括心脏毒性的有意义差异。两个臂中最常见的不良事件是脱发,恶心,腹泻和贫血。1,7

新闻稿
baar(瑞士),2024年12月2日 - 专门从事过敏原免疫疗法(AIT)的生物制药公司Stallergenes Greer今天宣布,人类使用委员会(CHMP)的欧洲药品机构(EMA)为现有的Incoptiation forPalforzia®的扩展提供了积极意见(花生))对幼儿(1至3岁)的治疗,并确认对花生过敏的诊断。如果欧盟委员会授予扩展指示,Palforzia®将成为EMA认可的口服免疫疗法(OIT)治疗,以治疗患有确认的花生过敏的幼儿。palforzia®旨在通过精心控制和监督的初始剂量升级,上剂量和维护来逐渐提高人体耐受少量花生(脱敏)的能力。随着禁忌症的调整,适应症的扩展将使治疗能够在更早的年龄开始,从而为幼儿及其家人提供机会,从而减少因意外暴露于花生过敏原而产生严重过敏反应的风险。1欧盟委员会正在审查CHMP建议,该建议负责授予欧盟中央营销授权。如果被授予,营销授权将涵盖全部27个欧洲成员国和三个欧洲经济区国家(冰岛,列支敦士登和挪威)。2024年7月,美国食品和药物管理局(FDA)批准了Palforzia®的扩展指示,用于幼儿。监管提交是基于第三阶段Poseidon的数据(P eanut o ral免疫疗法的研究,用于e Arly i Arly I tervention for d esensitizati的研究),该研究发表在2023年的《新英格兰医学杂志》上。该研究评估了Palforzia®在1至3岁之间的花生过敏儿童中的功效和安全性,满足了其所有主要和次要疗效终点,并证明了有利的安全性。2“Palforzia®的积极建议标志着花生过敏的年幼儿童以及对家人的重要一步。这个里程碑是建立在Stallergenes Greer对提供创新解决方案的长期致力下的基础上,为过敏患者的利益。随着Stallergenes Greer的努力,为扩大对Palforzia®的访问而努力,该公司仍致力于为患者需求量身定制的解决方案。具有全面的投资组合,其中包括用于食品过敏,片剂和液体舌下溶液的口服免疫疗法,以及用于呼吸和昆虫毒液过敏的皮下配方,Stallergenes Greer为个性化的,精确的基于精确的过敏反应剂免疫疗法铺平了道路。

新闻稿FDA接受...
Press release FDA accepts Biologics License Application for subcutaneous maintenance dosing of Leqembi® (lecanemab-irmb) in the US Stockholm, January 14, 2025 – BioArctic AB (publ) (Nasdaq Stockholm: BIOA B) today announced that the U.S. Food and Drug Administration (FDA) has accepted BioArctic's partner Eisai's Biologics License Application (BLA)用于LEQEMBI皮下自身注射器(SC-AI),用于每周维持毒素的静脉内治疗,以治疗轻度认知障碍(MCI)或轻度痴呆阶段的疾病阶段(集体称为早期AD)的患者接受阿尔茨海默氏病(AD)。Leqembi是唯一可以通过家庭管理选项提供皮下注射的便利性的FDA批准的抗淀粉样疗法。《处方药用户费用法案》(PDUFA)操作日期已设置为2025年8月31日。BLA基于Clarity AD(研究301)开放标签扩展(OLE)和观察到的数据建模的数据。如果Leqembi皮下维持剂量得到FDA的批准,则Leqembi将是唯一可以使用自动注射器(AI)在家中皮下施用的AD的治疗方法。预计注射过程平均为15秒。SC-AI 360 mg每周维护方案将允许完成两周静脉内(IV)起始阶段的患者,与FDA进行了严格的讨论,可以接受每周的剂量,这些剂量有望维持临床和生物标志物益处。Leqembi已在美国,日本,中国,英国和其他市场获得批准。SC-AI有望简单易于患者及其护理伙伴使用,并且可能会减少对IV施用的医院或输液现场就诊和护理护理的需求,这将使继续维护管理变得更加容易,并可能有助于进一步简化AD的治疗途径。在2024年11月,该待遇从欧洲药品局(EMA)的人类使用委员会(CHMP)提出了批准。 ---该信息通过下面的联系人的代理发布,于2025年1月14日在CET下方发布。 有关更多信息,请联系:Oskar Bosson,VP Communications和IR电子邮件:oskar.bosson@bioarctic.se,电话:+46 70 410 71 80 80在2024年11月,该待遇从欧洲药品局(EMA)的人类使用委员会(CHMP)提出了批准。---该信息通过下面的联系人的代理发布,于2025年1月14日在CET下方发布。有关更多信息,请联系:Oskar Bosson,VP Communications和IR电子邮件:oskar.bosson@bioarctic.se,电话:+46 70 410 71 80 80
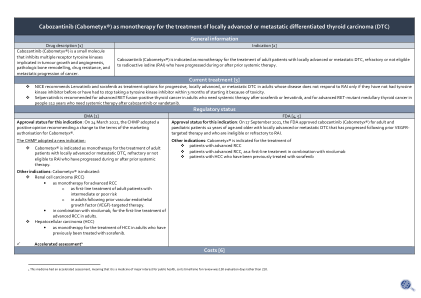
卡博替尼(Cabometyx®)作为单一疗法治疗局部晚期或转移性分化型甲状腺癌(DTC)
缩写:AE=不良事件,AJ=调整,ALT=丙氨酸氨基转移酶,AST=天冬氨酸氨基转移酶,C=比较器,CHMP=人用药品委员会,CI=置信区间,DTC=分化型甲状腺癌,EMA=欧洲药品管理局,ESMO-MCBS=欧洲肿瘤内科学会-临床获益量表,FDA=食品药品管理局,FM=最终临床获益等级,HCC=肝细胞癌,HR=风险比,I=干预,Int.=意向,ITT=意向治疗,MG=中位增益,n=患者人数,NA=不适用,NE=不可估计,NICE=英国国家健康与临床优化研究所,NR=未达到,OITT=意向治疗客观缓解率,ONJ=颌骨坏死, OS=总生存期,PE=主要终点,PFS=无进展生存期,PM=初步分级,PPE=掌跖红肿感觉异常,QoL=生活质量,RAI=放射性碘,RCC=肾细胞癌,RECIST=实体肿瘤疗效评价标准,RET=转染过程中重排,SAE=严重不良事件,ST=标准治疗,VEGF=血管内皮生长因子