XiaoMi-AI文件搜索系统
World File Search SystemClinvar

子宫内膜异位症和子宫肌症:长凳到床边
方法:患有腺瘤家族史的女性由于治疗异常的子宫出血而接受了子宫切除术。从外周血中分离出的基因组DNA在Illuina NextSeq 550系统上受到WES的影响,并利用扭曲综合外显子套件。生物信息学分析是在Genomize SEQ平台(https://seq.genomize.com)上进行的。候选基因从GWAS(基因组 - 韦德联合研究),NGS(下一代测序)和用于子宫腺癌及其紧密相关的子宫内膜异位症的功能研究都用于编译基因面板。平行滤波,以搜索基因面板中的稀有变体和WES数据中的新型变体。使用该平台的房屋等位基因频率来实现新奇人口的决定,该频率包括来自Turkiye的15,000个外显子组序列,患有不同的疾病。此外,根据疾病发病机理的功能相关性,优先考虑新的基因。使用ACMG(美国医学遗传学)指南以及临床数据库的致病性评分确定了用于疾病相关基因的数据库,而在新型变体评分中,使用了硅预测工具。对于新型候选人,仅致病性,可能的致病性和不确定意义的变体被视为腺瘤的候选变体。

开发与CacNA1A相关癫痫的临床试验途径:患者组织的观点
摘要:CACNA1A相关疾病是与Cacna1a基因中变体相关的罕见神经发育疾病。该基因编码P/Q-Type钙通道CAV2.1的α1亚基,该基因在大脑中全球表达,对于快速突触神经传递至关重要。与CACNA1A相关的神经系统疾病的广泛范围包括发育和癫痫性脑病,家族性偏瘫性偏头痛1型,多型性共济失调2型,脊髓灰质球共济失调6型,以及未分类的表现,以及发展性延迟,智力延迟,智力,自动化,自动化,以及语言幻象,以及语言谱。每种疾病的严重程度也高度可变。在功能丧失和功能获得的变体中,与CACNA1A相关的癫痫发作的频谱均广泛,包括缺席癫痫发作,意识改变的局灶性癫痫发作,普遍的音质持续性癫痫发作,强音性癫痫发作,状态性癫痫,癫痫持续性和女无水痉挛。此外,超过一半的CACNA1A相关癫痫是对当前疗法的难治性。迄今为止,在Clinvar中报道了将近1700个CACNA1A变体,其中400多个被列为致病性或可能的致病性,但具有限制于非临床或功能数据。鲁棒的基因型 - 表型研究以及变体对蛋白质结构和功能的影响尚未建立。结果,与CacNA1A相关癫痫的确定治疗选择很少。CACNA1A基金会已着手改变可用和有效治疗的景观,并改善与CacNA1A相关疾病(包括癫痫病)患者的生活质量。成立于2020年3月,该基金会建立了一个可靠的临床前工具箱,其中包括患者衍生的诱导多能干细胞和新型疾病模型,发起了临床试验准备计划,并组织了全球CACNA1A研究网络。该研究网络目前由60多名科学家和临床医生组成,他们致力于协作加速CACNA1A特异性治疗的道路,有一天可以治愈。
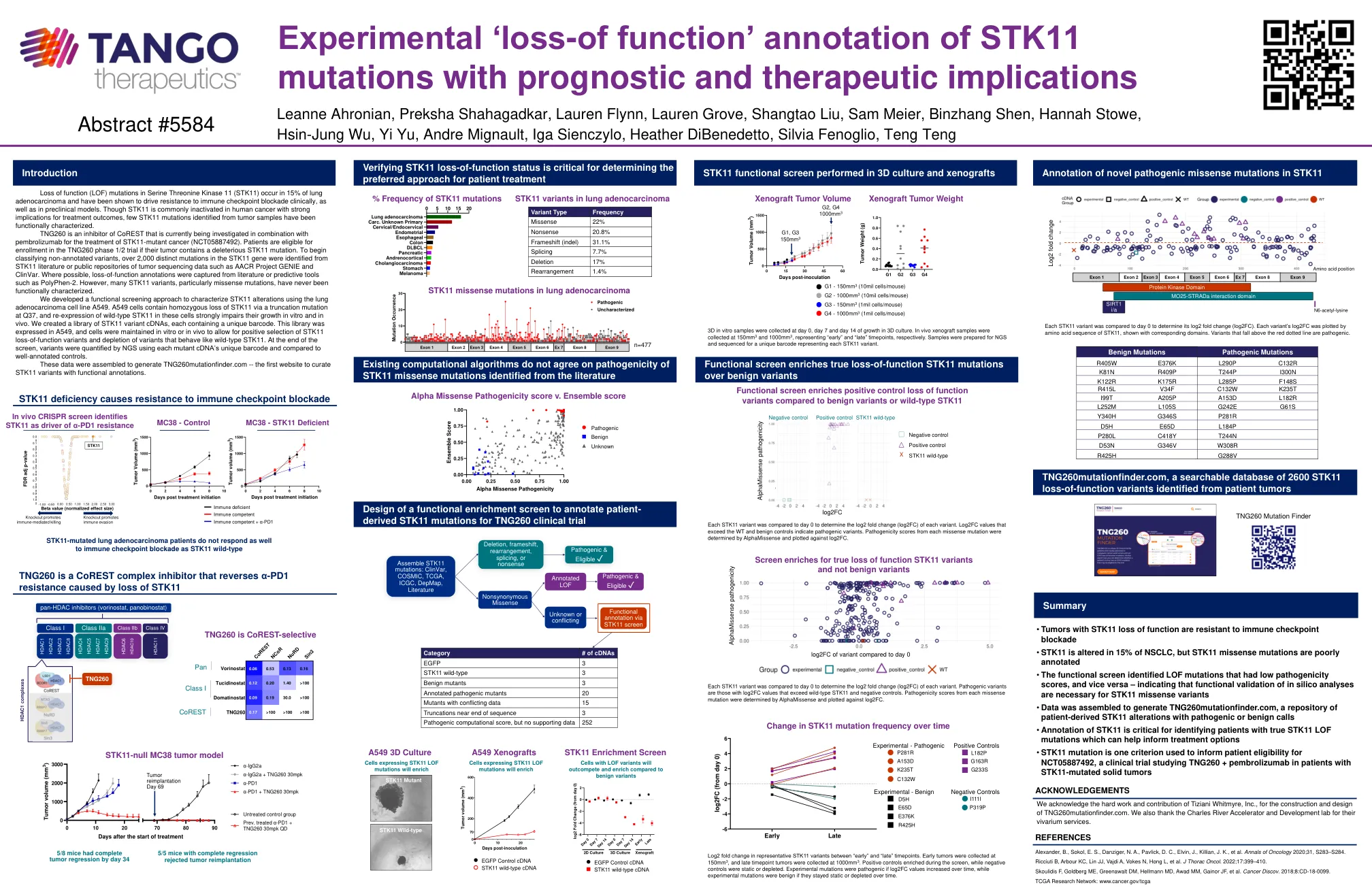
STK11的实验“功能丢失”注释...
丝氨酸苏氨酸激酶11(STK11)中功能(LOF)突变的丧失发生在15%的肺腺癌中,并且已被证明在临床上以及临床前模型中促进了对免疫检查点阻断的抗性。尽管STK11在人类癌症中通常被灭活,对治疗结果的影响很大,但是从功能上表征了从肿瘤样品中鉴定出的STK11突变。TNG260是Corest的一种抑制剂,目前正在研究与Pembrolizumab结合使用STK11-突变癌(NCT05887492)。患者有资格参加TNG260期1/2期试验,如果他们的肿瘤含有有害的STK11突变。为了开始对未经注销的变体进行分类,从STK11文献或肿瘤测序数据的公共存储库中鉴定出超过2,000个不同的突变,例如AACR Project Genie和Clinvar。在可能的情况下,从文献或诸如Polyphen-2之类的预测工具中捕获了功能丧失注释。但是,许多STK11变体,尤其是错义突变,从未在功能上表征。我们开发了一种功能筛选方法,使用肺腺癌细胞系A549表征STK11改变。A549细胞包含通过Q37处的截短突变纯合损失STK11,并且在这些细胞中重新表达了野生型STK11的表达,严重损害了它们在体外和体内的生长。我们创建了一个STK11变体cDNA的库,每个cdnas包含一个唯一的条形码。在屏幕末端,使用每个突变cDNA的独特条形码通过NGS对变体进行了定量,并将其与良好的对照对照进行了比较。该文库在A549中表达,并在体外或体内保持细胞,以允许对STK11功能丧失变体进行积极选择,并且耗尽了像野生型STK11的变体。这些数据被组装成生成TNG260Muntfinder.com-第一个策划具有功能注释的STK11变体的网站。

基因组报告示例
基因组坐标位置渗透载体表型基因覆盖g.8003996666delc(Chr17,grch38)外显子3高3个高note note note note note note note note 15倍变体解释:p.ala83valfsx84在CCDC40中的p.ala83valfsx84变异,先前在19个雄性和7型杂质的helel helesozygous and pc n hymozygous and pc contia和7 compio and syzygous and;在1个纯合受影响的亲戚中与疾病隔离(Becker-Heck 2011 PMID:21131974,Nakhleh 2012 PMID:22499950,Antony 2013 PMID:23255504,Zariwala,2013 PMID:23891469)。该变体已在gnomad(http://gnomad.broadinstitute.org)中鉴定出0.074%(860/1167354)的非欧洲欧洲染色体。但是,此频率足够低,可以与隐性等位基因频率保持一致。在Clinvar中也报道了这种变体(变体ID 31069)。该变体被预测会引起移架,从而改变蛋白质的氨基酸序列,从位置83开始,并导致下游的过早终止密码子84氨基酸。然后预测这种改变会导致截短或不存在的蛋白质。功能研究表明,CCDC40功能的丧失导致纤毛结构和运动异常(Becker-Heck 2011 PMID:21131974)。总而言之,该变体符合标准,该标准被归类为常染色体隐性原发性睫状运动障碍的致病性。ACMG/AMP标准应用:PVS1,PM3_VERYSTRONG,PM2_SUPPORTING,PP1。疾病信息:原发性睫状运动障碍是一种罕见的遗传病,在遗传上是异质的。它与复发性呼吸道感染,内脏异常定位以及不育有关。这是由于器官和组织衬里发现的纤毛和鞭毛的运动性异常。呼吸道感染,粘液清除率降低,鼻塞和慢性咳嗽始于幼儿,可能导致支气管扩张。Situs Inversus Totalis是所有内脏器官的镜像逆转,在40-50%的个体中发现。雌性运动障碍的雄性由于精子运动异常而经常是不育的,而患有这种疾病的女性有时可能是由于输卵管中的纤毛异常引起的。其他症状可能包括大脑中的复发性耳朵感染和脑积水。Pathogenic variants in CCDC40 contribute to 3-4% of primary ciliary dyskinesia (Medline Plus: https://medlineplus.gov/genetics/condition/primary-ciliary-dyskinesia, GeneReviews: https://www.ncbi.nlm.nih.gov/books/NBK1122).家族性和生殖风险疾病患病率(估计)载体频率(估计)生殖风险(估计)1/16000(https://medlineplus.gov/genetics/conditics/condition/primary-ciliary-ciliary-ciliary-dyskinesia)

免费和开明的同意
7。如何保存和使用我的遗传信息?数字文件不包含个人标识符。遗传数据只能由负责执行的医疗团队将其链接到患者。这项考试的结果是机密的。只有通过患者或其法定监护人的书面同意,他们才能被释放给第三方。我们承诺将Variants(VCF)数字文件存储至少2(两)年。最终可以使用该数字文件来考虑当前技术的限制。在遗传或生物信息学研究中,可能会继续匿名研究结果(即无法可逆地删除所有个人标识符),以通过Mendelics和Collaboring Instituctions改善过程和产品。将不可能通过将这些分析提供给患者产生的信息,因为将删除样本的个人标识符。同样,患者在这些研究中使用序列的使用也不能在财务上得到奖励,也无权获得这些分析产生的任何产品。这些分析的结果可以发表在医学和科学期刊上,并存放在美国国家卫生研究院(NIH)等遗传变异的公共银行中,以促进医学和科学知识的发展,并通过同样的疾病使其他人受益。

引用Corcuff M,Garibal M,Desvignes J-P,Guien C,Grattepanche C,Collod-BéroudG,MénoretE,Salgado D和BéroudC(2023),Prove
分类(Yorczyk等,2015; Kim等,2019),主要与ACMG AMP准则准则允许的主观性和不确定性程度有关。他们建议在解释过程中使用28个标准来区分:良性(可能是良性)的意义(VUS),可能是致病性和致病性变体。但是,仅在临床实践中获得这些标准的一部分,并且必须使用带注释的变体集合。为提供这样的资源,已经制定了各种倡议,包括Clinvar(Landrum等,2016),Clingen(Savatt等,2018),Varsome(Kopanos等,2019)和Intervar(Li and Wang,2017)。这些从专家和各种资源中收集数据,并可以为未报告的变体提供解释。然而,此自动化过程有时可能会产生不适当的结果,并且应谨慎查看数据。,如果我们专注于分类证据,一方面,最具挑战性的标准之一是PM1“位于突变的热点和/或关键和完善的功能域(例如,酶的活性位点),没有良性变化”,这是在报告的病例中使用的约10%(Amendola等人,2016年)。要提取此信息,自动化系统主要依赖Uniprot(Uniprot联盟。2017)和“ dbnsfp31a_interpro”,该数据库是DBNSFP(Liu等,2011; Liu等,2016)和Interpro(Mitchell等,2019)的域信息数据库,可在蛋白质家族,域,域和功能性点上包含有关蛋白质家族和功能性的信息。已经使用保守域数据库(CDD)(Marchler-Bauer等,2015)制定了其他计划,例如Subrvis分数(Gussow等,2016),旨在评估基因子区域对变体的不耐受性。通常,PM1标准与突变簇的功能区域的广泛视图相关联。然而,很难使用,因为这种聚类的定义不足和理解,如其在Vasome中的各种解释所示(Kopanos等人,2019年)和Intervar(Li and Wang,2017)。它也可能受到基因非人类疾病的兴趣和分类的变异次数的高度偏见。另一方面,最常用的证据是PM2/BA1/BS1“人口数据库中缺失的变异或等位基因频率太高,对于该疾病而言,据报道约有50%的病例(Amendola等人,2016年)。该标准的假设非常简单:如果已报告了普通人群频率高的变体,则不能是一种罕见的致病变异,否则该疾病的频率将更高;如果从未报道过变体,或者频率很低,则可能是一种罕见的致病变异。这些信息从大尺度基因组/外显子组测序项目中很大,大多数人从侏儒(Koch,2020年)或人口数据库中收集了这些信息,例如阿巴拉姆(巴西人人口)(Naslavsky等人)(Naslavsky等人,更大的Midder Midder eali Milder Elide Elide Elide Elide Elide Elide Elide Elide Elide Elide Elide Elide Elide Elide Elide eLDEL,202)人口)(Scott等,2016)。然而,人类进化不允许变异的基因组饱和,其中一些在遗传漂移引起的人群中非常罕见(Bach,2019)。的确,如果人口足够大,几代人几代人的失踪率很可能会导致其消失,而只有少数几代人将在人群中固定。因此,尽管人们认识到,从人类中出现了每一代人的50至100个变种,但这些事件中的大多数在进化过程中都丢失了,这解释了为什么我们的基因组中不存在所有中性替代。另一种观点是基于一个简单的假设,即78亿活着人类中每一个中的50至100从头变体都应该产生与生命兼容的每种核苷酸变化