XiaoMi-AI文件搜索系统
World File Search SystemCyclin

细胞周期蛋白依赖性激酶和罕见发育障碍 Pierre Colas
过去 30 年的大量研究已证实,依赖细胞周期蛋白的激酶 (CDK) 在发育过程中的许多分子和细胞过程中发挥着多种多样的重要功能。影响 CDK 或其激活细胞周期蛋白亚基的突变与多种罕见的人类发育障碍有关,这并不令人意外。本文回顾了这些最新发现,特别关注了已发现的突变及其已证实或假设的功能后果,这些后果可以解释人类的病理表型。本综述重点介绍了与人类疾病相关的新的、重要的 CDK 或细胞周期蛋白功能,并讨论了小鼠模型在揭示其中一些功能方面的不足之处。它解释了如何将人类遗传学与蛋白质组规模的相互作用数据库结合使用,以构建围绕 CDK 和细胞周期蛋白的调控网络。最后,它提倡使用这些网络来分析致病的 CDK 或细胞周期蛋白变体,以便了解蛋白质功能和致病机制。

细胞对CDK4抑制剂palbociclib的长期暴露会导致染色体像差
抽象的乳腺癌通常是由突变,细胞周期调节蛋白的变化和激活驱动的,包括视网膜细胞瘤肿瘤抑制蛋白(RB),细胞周期蛋白E和细胞周期蛋白依赖性激酶(CDKS),尤其是细胞周期蛋白D:CDK4/6复合物。目前有三种FDA认可的CDK4/6抑制剂(CDK4I)用于治疗乳腺癌。标准治疗方案是连续CDK4I治疗的21天,然后进行7天的停止期,然后重复28天的方案。我们询问了在7天CDK4I停止期间重新进入细胞周期的细胞会发生什么问题。使用含有视觉报道器内源性组蛋白2B和p27基因的RPE1细胞标记为EGFP和MCHERRY,我们用CDK4I,palbociclib处理了1至42天的细胞,跨越了临床暴露,通过药物释放(PARKOPIC)的释放,我们发现了临床暴露的时间。在微核和多核细胞中,已重新进入细胞周期。峰值染色体畸变发生在14到35天之间,这个时间跨越了临床给药方案。这些观察结果提出了有关循环患者在CDK4抑制剂中的循环和关闭的潜力,可能会导致染色体细胞对肿瘤细胞的总体变化,从而在7日临床上产生临床的临床范围,从而使肿瘤循环逐渐增加。关键词palbociclib,CDK4/6抑制剂,CDKN1B在美国女性引入,乳腺癌仍然是癌症的最常见形式,也是癌症第二常见的死亡原因[1]。乳腺癌通常是由突变,细胞周期调节蛋白的变化和激活驱动的,包括视网膜细胞瘤肿瘤抑制蛋白(RB),细胞周期蛋白E和细胞周期蛋白依赖性激酶(CDKS)[2,3]。以前,Cyclin d:CDK4复合物被认为通过低磷酸化灭活RB [4,5];然而,我们实验室和其他人的最新证据现在表明,在细胞周期的G1早期,细胞周期蛋白D:CDK4仅定量单磷酸化RB,并且14个单磷酸化的RB同工型每个选择性地结合细胞靶标[6-8]。 相反,限制点的细胞周期蛋白E:CDK2复合物的激活通过高磷酸化进行初始RB灭活,触发E2F转录因子的释放,进展为G1晚期,然后进入S相[6,7]。 尚不清楚Cyclin d:Cdk4的非RB靶标在G1早期以驱动细胞周期进展,但很明显,连续的细胞周期蛋白D:CDK4活性是许多乳腺癌(包括乳腺癌)早期G1细胞周期进展的必需驱动器[9]。 FDA批准了第一个CDK4抑制剂(CDK4I),Palbociclib(IBRANCE),是2015年的突破性治疗方法,用于治疗雌激素受体阳性(ER+)乳腺癌[10]。 在2017年进行了此跟踪,并批准了CDK4I在ER+/HER2阴性乳腺癌中使用[11]。 鉴于细胞周期蛋白D:CDK4在驱动癌症中的重要性,现在有三种FDA批准的抑制剂[12]也就不足为奇了。 标准的CDK4I治疗连续21天与抗雌激素药物结合使用,然后进行7天的戒烟,然后重复28以前,Cyclin d:CDK4复合物被认为通过低磷酸化灭活RB [4,5];然而,我们实验室和其他人的最新证据现在表明,在细胞周期的G1早期,细胞周期蛋白D:CDK4仅定量单磷酸化RB,并且14个单磷酸化的RB同工型每个选择性地结合细胞靶标[6-8]。相反,限制点的细胞周期蛋白E:CDK2复合物的激活通过高磷酸化进行初始RB灭活,触发E2F转录因子的释放,进展为G1晚期,然后进入S相[6,7]。尚不清楚Cyclin d:Cdk4的非RB靶标在G1早期以驱动细胞周期进展,但很明显,连续的细胞周期蛋白D:CDK4活性是许多乳腺癌(包括乳腺癌)早期G1细胞周期进展的必需驱动器[9]。FDA批准了第一个CDK4抑制剂(CDK4I),Palbociclib(IBRANCE),是2015年的突破性治疗方法,用于治疗雌激素受体阳性(ER+)乳腺癌[10]。在2017年进行了此跟踪,并批准了CDK4I在ER+/HER2阴性乳腺癌中使用[11]。鉴于细胞周期蛋白D:CDK4在驱动癌症中的重要性,现在有三种FDA批准的抑制剂[12]也就不足为奇了。标准的CDK4I治疗连续21天与抗雌激素药物结合使用,然后进行7天的戒烟,然后重复28

Atirmociclib (PF-07220060) 和 PF-07104091 是首批
参考文献: 1. Choi YJ, Anders L. 通过细胞周期蛋白 D 依赖性激酶进行信号传导。Oncogene。2014 年 4 月 10 日;33(15):1890-903。doi: 10.1038/onc.2013.137 2. VanArsdale 等人。针对细胞周期蛋白 D-CDK4/6 轴进行癌症治疗。Clin Cancer Res。2015 年 7 月 1 日;21(13):2905-10。doi: 10.1158/1078-0432.CCR-14-0816 3. Fassl 等人。CDK4 和 CDK6 激酶:从基础科学到癌症治疗。Science。2022 年 1 月 14 日;375(6577):eabc1495。 doi: 10.1126/science.abc1495

JAC1 靶向 YY1 介导的 JWA/p38 MAPK 信号传导以抑制 TNBC 中的增殖并诱导细胞凋亡
以剂量依赖性方式调节,同时,JAC1处理MDA-MB-231和SUM1315细胞24h后,Bax和cleaved-caspase3均上调(图3E)。细胞周期测定显示,用5 mM JAC1处理两种TNBC细胞24h后,G1期细胞增多,S期细胞明显减少。此外,JAC1对细胞周期阻滞的影响在SUM1315细胞中比在MDA-MB-231细胞中更敏感(图3F-I)。当用JAC1处理TNBC细胞24h时,p21和CDK6(不是CDK4)的表达增加,但cyclin D1被明显抑制(图3J)。数据还显示JAC1对p21的作用比对cyclin D1更敏感。综上所述,这些结果表明 JAC1 在细胞凋亡和细胞周期停滞中发挥双重作用。
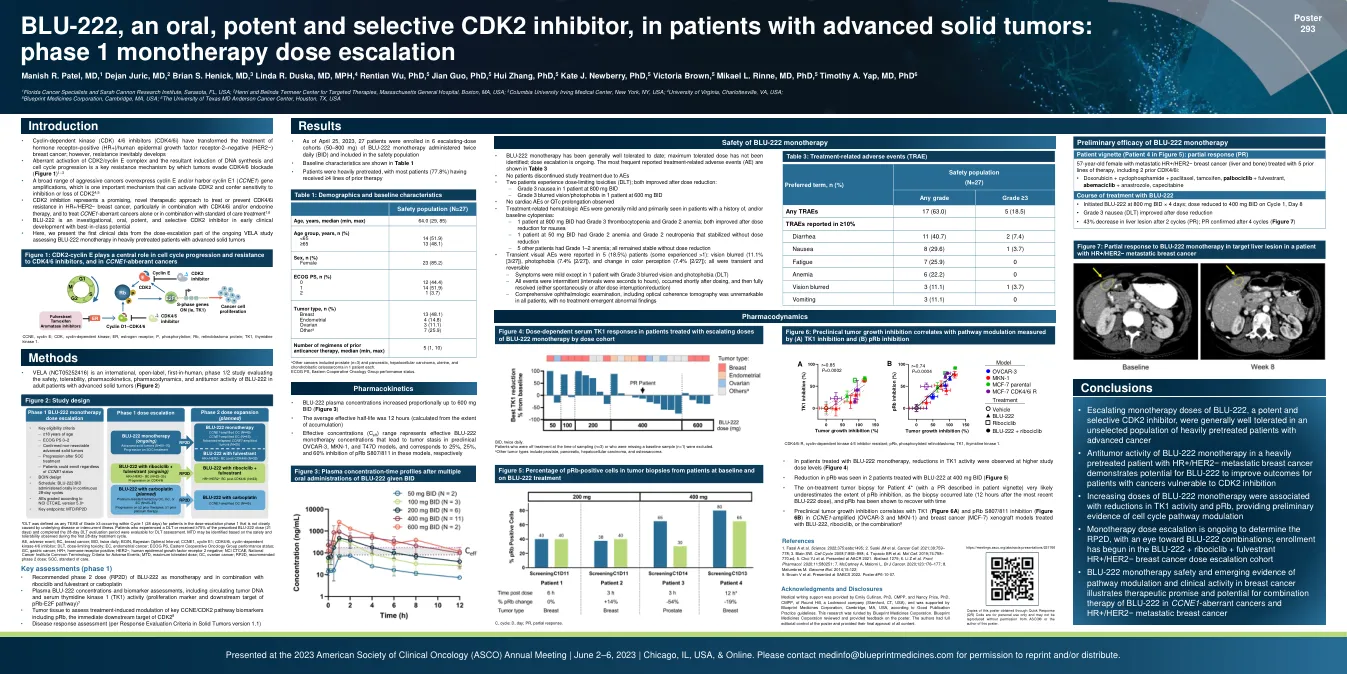
BLU-222 是一种口服、强效且选择性的 CDK2 抑制剂,...
• 细胞周期蛋白依赖性激酶 (CDK) 4/6 抑制剂 (CDK4/6i) 改变了激素受体阳性 (HR+)/人类表皮生长因子受体 2 阴性 (HER2-) 乳腺癌的治疗;然而,耐药性不可避免地会产生 • CDK2/细胞周期蛋白 E 复合物的异常激活以及由此导致的 DNA 合成和细胞周期进展的诱导是肿瘤逃避 CDK4/6 阻断的关键耐药机制(图 1)1-3 • 多种侵袭性癌症过度表达细胞周期蛋白 E 和/或携带细胞周期蛋白 E1(CCNE1)基因扩增,这是一种可激活 CDK2 并赋予对 CDK2 抑制或缺失的敏感性的重要机制 4,5 • CDK2 抑制代表了一种有前途的新型治疗方法,可用于治疗或预防 HR+/HER2-乳腺癌中的 CDK4/6i 耐药性,特别是与 CDK4/6i 和/或内分泌疗法相结合,以及单独或与标准治疗方法相结合治疗 CCNE1 异常癌症 1,6 • BLU-222 是一种在研的、口服的、强效的、选择性的 CDK2 抑制剂,处于早期临床开发阶段,具有同类最佳的潜力 • 在这里,我们介绍了第一个正在进行的 VELA 研究剂量递增部分的临床数据,该研究评估了 BLU-222 单药治疗接受过大量治疗的晚期实体瘤患者

非规范CDK4信号传导在A ...
引言内源性胰腺β细胞质量和功能的增加将解决胰岛素缺乏症患者造成的危害。营养暴露,例如高血糖(1),高脂肪饮食(2、3)和其他营养过多的范式(4)通过有丝分裂的输入促进β细胞增殖,从而在下流信号上融合以横向流动,从而横向传播细胞周期的G1/s(3,5,5,6)。另一方面,通过β细胞死亡(7)和去分化(8,9)可能会损失胰岛素分泌能力;一些数据表明,人类2型糖尿病(T2D)中β细胞质量的减少可能已经高估了(10)。尽管如此,β细胞再生场中的关键障碍是增加了替补的策略也可能导致去分化(8、9、11)。胰岛素受体底物– 2(IRS-2)的全身缺失导致T2D样综合征由于β细胞功能降低和肿块而表现出明显的胰岛素耐药性(12,13)。通过抑制键β细胞因子PDX1(9,14),在该模型中远端胰岛素信号通路成员叉子盒蛋白O1(FOXO1)的因素推导。irs2 - / - β细胞也因细胞周期蛋白D2的诱导降低而对葡萄糖的增殖反应受损,并且恢复细胞周期蛋白D2丰度挽救了增殖到正常水平(15)。Cyclin D2,是产后β细胞膨胀的驱动因素和胰岛素抵抗的β细胞补偿(1,16-20),与Cyclin依赖性
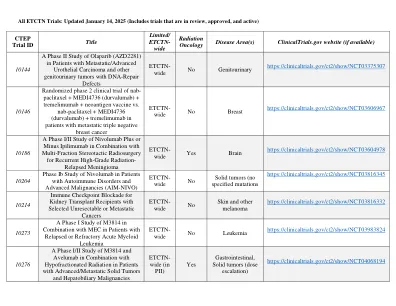
NCTN试用组合截至2025年1月,所有ETCTN癌症临床试验
曲妥珠单抗Deruxtecan(DS-8201A)与Azenosertib(Zn-C3)在HER2-表达/扩增的细胞周期蛋白E-扩增的胃/胃食管治疗疗法和其他实体瘤与HER2表达

海报#A023
• CDK1 activation is monitored by Cyclin B phosphorylation (CyclinB-pS126) • Histone H3 phosphorylation (H3-pS10) in EdU-incorporating cells marks premature mitotic entry in S-phase • Lunresertib + WEE1i induces faster and stronger CDK1 activation and premature mitosis in CCNE1 -high cells compared to parental counterparts以最小的单位活动活性在任何背景

1。细胞繁殖
§CDKS水平通常是恒定的。§CDK是不活跃的。§cdks通过与细胞周期蛋白结合并受磷酸化和去磷酸化的调节而激活。§CDK将受到G 1,G 2和M检查点的调节。Cyclin-CDK复合物的一个例子是促进因子(MPF,也称为有丝分裂因子 - 促进因子或M期促进因子),该因子由调节亚基-Cyclin b和催化亚基 - Cyclin依赖性激酶(CDK1,CDC2或P34 KINS)组成,该型和P34 KIN酶是刺激的。MPF通过磷酸化有丝分裂过程中所需的多种蛋白质来促进从G 2期进入有丝分裂的入口。MPF在G 2的末尾被磷酸酶酶激活,该酶消除了较早添加的抑制性磷酸组。外部信号生长因子是某些刺激其他细胞分裂的人体细胞释放的蛋白质。密度依赖性抑制 - 拥挤的细胞停止分裂时的现象。锚定依赖性 - 何时必须将细胞分开的现象必须连接到底层。锚固与质膜蛋白有关。

解释了CDK介导的细胞周期控制中的冗余:统一连续性和定量模型
摘要:在真核生物中,Cyclin依赖性激酶(CDKS)是DNA复制和有丝分裂的必需的,并且在整个细胞周期中,依次激活了不同的CDK-循环蛋白复合物。普遍认为,特定的复合物需要遍历G1中细胞周期的承诺,并分别促进S期和有丝分裂。因此,根据一个流行的模型,几十年来一直占据了领域的流行模型,在细胞周期的每个阶段,针对不同底物的独特CDK – cyclin compleces固有的特定座位生成了事件的正确顺序和时间。但是,编码细胞周期蛋白和CDK的基因敲除的结果不支持此模型。通过许多最近的工作验证的替代性“定量”模型表明,CDK活性的总体水平(具有相反的磷酸酶输入)决定了S期和有丝分裂的时间和顺序。我们通过建议将细胞周期分为离散阶段(G0,G1,S,G2和M)的细分被过时且有问题,从而进一步采用了该模型。相反,我们恢复了细胞周期的“连续性”模型,并提出与定量模型的结合更好地定义了理解细胞周期控制的概念框架。