XiaoMi-AI文件搜索系统
World File Search SystemF508del

Error 500 (Server Error)!!1500.That’s an error.There was an error. Please try again later.That’s all we know.
囊性纤维化(CF)是由CF跨膜电导调节剂(CFTR)基因突变引起的罕见疾病。在法国,超过80%的CF患者是纯合或杂合状态中F508DEL突变的载体。f508del导致过早降解但其他功能性蛋白质的合成。现在有很大的兴趣实施CFTR校正器,旨在营救顶部膜中的F508DEL。有些处于临床状态,但仍处于适度效率。需要使用相关的CF动物模型来对这些新化合物进行临床前研究。CF小鼠模型表现出几种相关的疾病,但无法模仿严重的肺部疾病:因此,已经开发了其他模型作为猪和雪貂中的CF。这两个物种都表现出有趣的气道障碍,但它们需要专门的设施和管理,这些设施和管理不容易获得,并带来了高昂的成本。此外,由于其剧烈的肠道疾病,只有少数动物达到成年年龄。最近,已经开发出了一条敲除(KO)大鼠,该大鼠在人类受试者中发现了许多CF表型的特征,包括气道粘液产生,气管发育,气道表面和周围液体液深,鼻粘液,牙齿,牙齿,牙齿,牙齿牙齿的缺陷以及VAS deferens的参与。大鼠是CF肺部疾病的有吸引力的模型,因为与小鼠不同,但与人类相似,它们在整个气管中都会形成广泛的粘膜下腺,直至支气管水平。尽管如此,CFTR-KO大鼠不允许研究校正器。F508DEL大鼠将通过Nantes的Trip Platform实现。因此,我们旨在使用F508DEL突变和单链寡脱氧核苷酸(SSODN)开发CF大鼠模型,该方法将与群集的定期散布的短palindromic重复(CRISPR)共同注入。这种新模型将使我们能够加深对由F508DEL突变引起的CF生理病理学的知识,尤其是关于肺部疾病和其他CFTR相关异常的知识。此外,F508DEL大鼠模型将使我们能够测试校正器对另一种模型中CF病理的影响,从而提供了有关其功效,代谢和毒性的有价值的补充数据。这种F508DEL大鼠模型将是世界上第一个模型,并构成了CF研究的重大进展。

PNA 纳米粒子介导的囊性纤维化的体内矫正
囊性纤维化 (CF) 是由 CF 跨膜传导调节器 (CFTR) 基因突变引起的。我们试图通过系统性递送肽核酸基因编辑技术(由生物相容性聚合物纳米颗粒介导)来纠正 F508del CF 致病突变引起的多器官功能障碍。我们在气液界面生长的 F508del 小鼠的原代鼻上皮细胞中证实了体外表型和基因型修饰,并在静脉内递送后在 F508del 小鼠体内证实了表型和基因型修饰。体内治疗导致上皮细胞中 CFTR 功能部分增强(通过原位电位差和 Ussing 室测定测量)以及气道和胃肠道组织中的 CFTR 得到纠正,并且没有高于背景的脱靶效应。我们的研究表明系统性基因编辑是可能的,更具体地说,静脉内递送旨在纠正 CF 致病突变的 PNA NP 是改善多个受影响器官中 CF 的可行选择。

治疗引起囊性纤维化的剪接突变的基因手术
当前的工作符合 CF 领域正在进行的努力,旨在满足所谓“最后 10%”的高度未满足的医疗需求,即 pwCF,根据其特定的基因型,这些患者不适合 HEMT 并且处于前调节剂时代。除了严重的错义突变之外,这些基因型还包括剪接、插入或缺失 (indel) 或无义突变,从机制上来说,预计这些突变不会对任何当前或未来的调节剂疗法产生反应。为了解决这一未满足的医疗需求,CF 领域努力研究基因添加和基因编辑方法(见表 1)。事实上,自从 HEMT 最常见的突变 F508del 以及门控和残留功能突变获得临床批准以来,药物难治性突变一直是研究的重中之重(见表 1)。 c.3718-2477C>T 是一种残留功能突变,携带此类突变的 pwCF 现在有资格获得美国批准的 CFTR 调节剂(https://www.fda.gov/)。6 在欧洲,只有携带突变与 F508del 等位基因结合的 pwCF 才有资格获得 Symkevi(tezacaftor/ivacaftor)

小鼠体内编辑肺干细胞以实现持久的基因校正
体内基因组校正有望产生持久的疾病治疗方法;然而,有效的干细胞编辑仍然具有挑战性。在这项研究中,我们证明优化的肺靶向脂质纳米颗粒 (LNP) 能够在干细胞中进行高水平的基因组编辑,从而产生持久的反应。在可激活的 tdTomato 小鼠中静脉注射基因编辑 LNP 可实现 >70% 的肺干细胞编辑,并在 >80% 的肺上皮细胞中维持 tdTomato 表达 660 天。解决囊性纤维化 (CF),NG-ABE8e 信使 RNA (mRNA) – sgR553X LNPs 介导 >95% 的囊性纤维化跨膜传导调节器 (CFTR) DNA 校正,恢复原发性患者支气管上皮细胞中的 CFTR 功能,相当于 Trikafta 治疗 F508del,校正肠道类器官并校正 CF 小鼠 50% 肺干细胞中的 R553X 无义突变。这些发现引入了 LNP 支持的组织干细胞编辑,用于疾病修饰基因组校正。G

CAGE 测序揭示了活化囊性纤维化巨噬细胞中 CFTR 依赖的 I 型 IFN 信号失调
强烈的、无法缓解的气道炎症反应会导致囊性纤维化 (CF) 患者的破坏性肺部疾病。巨噬细胞免疫功能失调可能是控制 CF 肺部疾病进展的一个关键方面,但其潜在机制尚不完全清楚。我们使用 5′ 端为中心的转录组测序来分析铜绿假单胞菌 LPS 激活的人类 CF 巨噬细胞,结果显示 CF 和非 CF 巨噬细胞在基线和激活后部署了截然不同的转录程序。这包括与健康对照相比,激活的患者细胞中 I 型 IFN 信号反应明显减弱,但在患者细胞中使用 CFTR 调节剂进行体外治疗以及通过 CRISPR-Cas9 基因编辑来纠正患者来源的 iPSC 巨噬细胞中的 F508del 突变后,这种反应是可逆的。这些发现表明,人类 CF 巨噬细胞中存在以前未被发现的免疫缺陷,这种缺陷依赖于 CFTR,并且可以通过 CFTR 调节剂逆转,从而为寻找 CF 中的有效抗炎干预措施提供了新的途径。

囊性纤维化患者细胞中的 P.F508del 编辑
基因组编辑方法的发展为基于病因的遗传性疾病治疗方法的开发创造了新的机遇。在这里,我们证明 CRISPR/Cas9 可以纠正囊性纤维化 (CF) 患者来源的 CFTE29o- 细胞和诱导多能干细胞 (iPSC) 中 CFTR 基因的 p.F508del 突变。我们使用了 Cas9、sgRNA 和 ssODN 的几种组合,并测量了内源性 CFTR 基因和含有 p. F508del 突变 CFTR 基因座的共转染质粒的编辑效率。CFTE29o- 细胞中 CFTR 基因中的非同源末端连接 (NHEJ) 频率从等位基因的 1.25% 到 2.54% 不等。内源性 CFTR 基因座中最好的同源定向修复 (HDR) 频率为等位基因的 1.42%。在 iPSC 中,CFTR 基因中的 NHEJ 频率从等位基因的 5.5% 到 12.13% 不等。HDR 效率最高的是等位基因的 2.38%。我们的结果表明,在 CF 患者来源的 iPSC 中使用 CRISPR/Cas9 进行 p.F508del 突变编辑是一种相对罕见的事件,应进行后续细胞选择和培养。

santos_jcf_2022-crispr.pdf
在CFTR基因中已经识别出来,其中8%是非理性变体[2,3]。CFTR基因中第二大最普遍的废话变体是W1282X(C.3846G> A),占全球等位基因的1.2%[2]。W1282X-CFTR变体生成过早的终止密码子(PTC),该密码子(PTC)导致胡说八道介导的衰减(NMD),因此产生了很少或没有全长的CFTR蛋白[3]。在过去的十年中,食品药品监督管理局(FDA)和欧洲药品局(EMA)批准了四种不同的CFTR蛋白模型,以治疗CF的根本原因:一个增强剂(VX-770),这些原因(VX-770)增加了质量膜上的通道开放概率,并对97个不同的变化群和三个不同的变量(VX-809)(Vx-809,VX)(Vx)(Vx)(Vx)(Vx)(Vx)(Vx)(Vx)(Vx)(Vx)(Vx)(Vx)(Vx-880)(Vx)(Vx-880)(Vx)(Vx-8809), VX-445)改善了最普遍的CF变体F508DEL的蛋白质折叠和效果[4,5]。总共意味着约85%的CF患者有潜在的治疗方法。但是,由于这些药物直接作用于CFTR蛋白,因此它们不适合治疗患有W1282X和其他PTC变体的CF患者。

囊性纤维化的新治疗方法的有效性和价值
在过去十年中,美国食品药品监督管理局(FDA)批准了几种直接调节异常CFTR蛋白的新型药物。药物靶向特定类别的CFTR基因突变。FDA于2019年10月21日批准了Trikafta(Elexacaftor/ tezacaftor/ ivacaftor,tezacaftor/ ivacaftor,vertex Pharmaceuticals)的口服三重疗法,用于2019年10月21日,用于在12岁及以上的患者中治疗CF,年龄至少为1份F508DEL突变的患者。大约90%的CF患者存在此突变。其他3种FDA批准的调节疗法是Kalydeco(Ivacaftor),Orkambi(Lumacaftor/Ivacaftor)和Symdeko(Tezacaftor/Ivacaftor)。临床和经济综述研究所(ICER)进行了系统的文献综述和成本效益分析,以评估这些CFTR调节剂疗法的健康和经济成果。可以在ICER的网站上获得ICER系统文献搜索和协议的完整详细信息,以及经济评估的方法和模型结构。在这里,我们在2020年8月27日在加利福尼亚技术评估论坛的一次公开会议上与关键利益相关者介绍了我们的发现和政策讨论的重点。详细报告可用
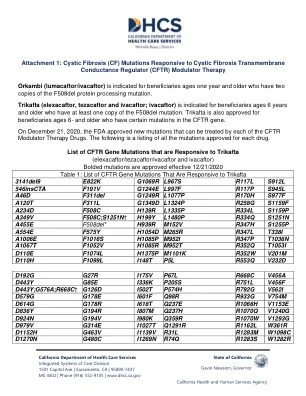
附件1:囊性纤维化(CF)突变,对囊性纤维化跨膜
(ELEXACAFTOR/TEZACAFTOR/IVACAFTOR和IVACAFTOR)粗体突变被批准有效12/21/2020表1:响应Trikafta 3141del9 E822K G1069R L967S R967S R117L L967S R117L S912L S912L 546INSCA的CFTR基因突变列表R117P S945L A46D F311DEL G1249R L1077P R170H S977F A120T F311L F311L G1349D L1324P R258G S1159F S1159F A234D A234D F508C H139R L1335P L1335P R334L S1159P; H199Y L1480P R334Q S1251N A455E F508DEL* H939R M152V R347H S1255P A554E F575Y H1054D H1054D A1067T F1052V H1085R M952T R352Q T1053I D110E F1074L H1375P M1101K R352W V201M D110H F1099L F1099L I148T I148T P553Q V232D D192G D192G G27R I117R I117R I117R I117R I117R I117R I117R RAR D443Y G85E I336K P205S R751L V456F D443Y; G576A; R668C; R668C†G126D I502T P574H R792G V562I V562I D579G D579G G178E D578E I601F i601f i601f i601f Q98R R933G V733G V733G V733G V754M D614M D614M D614MD614MD614MD614MD614MD614MD614MD614MD614MD614MD614MD614 d614 d614m d614m d614 d614 d614 Q237E R1066H V1153E D836Y G194R I807M Q237H R1070Q V1240G D924N G194V I980K Q359R R1070W V1293G D979V G314E I1027T Q1291R R1162L W361R D1152H G463V I1139V R31L R1283M W1098C D1270N G480C I1269N R74Q R1283S W1282R

肠道杀菌剂在囊性纤维化小鼠模型中通过丙酸调节炎症,全身细胞因子和微生物生态学
囊性纤维化(CF)的抽象人员从早期开始,显示出肠道微生物组营养不良的部分,部分原因是菌群的相对丰度降低。杀菌剂是肠道短链脂肪酸丙酸的主要生产国。我们在这里证明了囊性纤维化跨膜诱导调节剂缺陷(CFTR - / - )CACO-2肠上皮细胞对丙酸丙酸酯的抗炎作用有反应。此外,杀菌剂分离株抑制了IL-1β诱导的CFTR - / - CACO-2肠上皮细胞的炎症反应,并以丙酸依赖性方式进行。从患有囊性纤维化的婴儿中引入细菌质的粪便中,CFTR F508DEL小鼠的肠道导致粪便中丙酸的丙酸较高,并减少了几种全身性炎性细胞因子。杀菌剂补充剂还降低了大肠杆菌的粪便相对丰度,表明这两个微生物之间的潜在相互作用与以前的临床研究一致。与野生型(WT)菌株相比,在小鼠模型中,对于小鼠模型中的细菌丙酸酯突变体,促炎性细胞因子KC较高,这两种菌株的绝对丰度没有显着差异。总而言之,我们的数据表明了菌孢子源性的潜在多重作用在调节系统性和气道炎症中,并介导了婴儿和儿童CF的肠道生态学。菌孢菌的作用及其产生的丙酸酯可能有助于解释CF中观察到的肠肺轴,并可以指导益生菌的发展以减轻CF患者的全身性和气道炎症。