XiaoMi-AI文件搜索系统
World File Search SystemFriedreich

Friedreich ataxia(FA)情况说明书
心脏异常发生在大约75%的FRDA患者中,但严重程度差异很大。FRDA中最常见的异常是肥厚性心肌病,心脏肌肉肿大会缩小心脏中充满血液的腔室,降低泵送能力并导致心力衰竭。对于FRDA的人来说,与心脏病专家进行定期检查是个好主意。心脏病专家将选择最适合每位患者的治疗方法。定期监测心脏功能和节奏将使他们能够尽早治疗药物或起搏器。药物可用于减少心脏的工作,减慢心率或减少异常心律的机会。

Friedreich共济失调儿童的临床试验
Friedreich的共济失调研究联盟(FARA)认为,当预计最大的治疗益处时,必须将FA患者尽可能年轻。因此,FARA希望确保在临床试验的早期包括FA的儿童,并因此迅速获得安全,有效的治疗方法。启用小儿试验需要提起者,调查员,足总社区和监管机构之间的早期协作讨论。FARA已开发此文件,以提供有关儿童疾病课程的关键信息,儿科试验的监管观点和要求,生物标志物和临床评估方法以及临床试验设计,以及FA对儿童参与临床试验的看法。法拉在制定小儿试验计划时渴望支持行业伙伴,包括参与与监管机构的讨论,从很早就参与开发过程中,因为Fara确信,我们可以共同使FA儿童既可以纳入临床试验,从而大大增加了这些试验的成功率,并提供了这些试验的可能性,并提供了待遇,并提供了可靠的儿童和有效的治疗方法。

NFL和PNFH在Friedreich的Ataxia中增加了
抽象的目的是评估弗里德里希共济失调患者血清中神经退行性生物标志物的神经丝。在99例遗传确认的弗里德里希共济失调的患者中,神经丝(NFL)和重链(PNFH)的单分子阵列测量值。NFL/PNFH血清水平与疾病严重程度,疾病持续时间,年龄,发病年龄和GAA重复长度的相关性。在对照组中,NFL的中值NFL水平为21.2 pg/mL(范围3.6-49.3),而弗里德里希(Friedreich)的共济失调(p = 0.002)中的结果为21.2 pg/ml(范围3.6-49.3),26.1 pg/ml(0-78.1)。PNFH水平为23.5 pg/ml(13.3-43.3),在弗里德里希共济失调(P = 0.0004)中,对照组中的PNFH水平为92 pg/ml(3.1-303)。NFL水平显着升高,但48岁及48岁以上的患者(p = 0.41)(p = 0.41)。在纵向评估中,在基线测量后2年重复进行采样,NFL水平没有差异。NFL水平与GAA1重复长度(r = -0.24,p = 0.02)呈媒体相关,但与疾病的严重程度无关(r = −0.13,p = 0.22),疾病持续时间(r = −006,p = 0.53)或年龄(r = 0.05,p = 0.05,p = 0.62)。结论NFL和PNFH的血清水平升高,弗里德里希共济失调,但与健康对照的差异随着年龄的增长而降低。需要长期的纵向数据探索这是否反映了受影响更严重的个体的早期死亡或随着年龄的增长而减慢神经退行性过程的选择偏见。在一项在2年的随访中进行的试点研究(与表明治疗效果的生物标志物相关的时期),我们发现NFL水平是稳定的。

专制和环境之间的联系... -Iris
Bolotta,A.,Pini,A.,Abruzzo,P.M.,Ghezzo,A.,Modesti,A.,Gamberi,T。等。(2020)。弗里德里希共济失调中补充三烯醇的影响:氧化应激病理学的模型。实验生物学和医学,245(3),2012-212 [10.1177/1535370219890873]。

基因治疗和基因编辑治疗弗里德赖希共济失调的优势和局限性
弗里德赖希共济失调 (FRDA) 是一种遗传性多系统疾病,主要由 frataxin (FXN) 基因内含子 1 中的 GAA 过度扩增引起。这种扩增突变在转录上抑制了 FXN,FXN 是一种线粒体蛋白,是铁代谢和线粒体稳态所必需的,导致神经退行性和心脏功能障碍。目前,FRDA 的治疗方案集中于通过药物干预改善线粒体功能和增加 frataxin 表达,但在临床试验中无法有效延缓或预防神经退行性病变。最近在 FRDA 动物和细胞模型中对体内和体外基因治疗方法的研究展示了其作为 FRDA 一次性疗法的前景。在本综述中,我们概述了 FRDA 基因治疗的当前和新兴前景,特别关注 CRISPR/Cas9 介导的 FXN 基因编辑作为恢复内源性 frataxin 表达的可行选择的优势。我们还评估了造血干细胞和祖细胞中的体外基因编辑作为潜在的自体移植治疗选择的潜力,并讨论了其在解决 FRDA 特定安全问题方面的优势,以实现临床转化。

在弗里德里希共济失调中人类frataxin蛋白的遗传变异和结构建模的计算机分析
摘要:弗里德里希(Friedreich)的共济失调(FRDA)是最普遍的遗传性共济失调形式,以渐进的运动性共济失调,振动敏感性的丧失和骨骼畸形为标志,严重影响了日常功能。迄今为止,唯一可用于治疗FRDA的药物是最近获得FDA批准的Omaveloxolone(Skyclarys®)。负责细胞内铁稳态调节的人frataxin(FXN)基因内的错义突变与FRDA发育有关。这些突变会诱导FXN功能障碍,促进线粒体铁的积累并增强氧化应激,最终触发神经元细胞死亡途径。这项研究合并了来自文献和数据库搜索的226个FXN遗传变异,并只有18个先前表征。预测分析表明,FXN突变的有害和不稳定预测的发生率显着,主要影响对蛋白质功能至关重要的保守残基。此外,构建了人类FXN的准确,全面的三维模型,是生成遗传变异I154F和W155R的基础。这些变体的严重临床意义,进行了分子动力学(MD)模拟,在其N末端段中揭示了灵活性和基本动态变化,其中包括FXN42,FXN56和FXN78领域的蛋白质成熟。因此,我们的发现表明在I154F和W155R突变引起的FXN42,FXN56和FXN78域中的潜在相互作用曲线干扰,与现有文献保持一致。
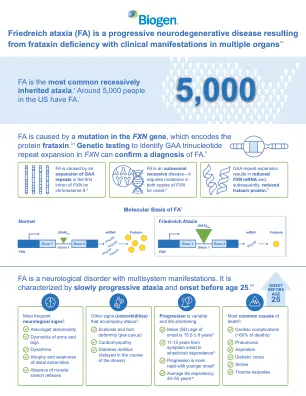
弗里德赖希共济失调 (FA) 是一种由 frataxin 缺乏引起的进行性神经退行性疾病,临床表现涉及多个器官 1
ARE,抗氧化反应元件;ATP,三磷酸腺苷;DNA,脱氧核糖核酸;FA,弗里德赖希共济失调;GAA,鸟嘌呤腺嘌呤腺嘌呤;ISC,铁硫簇;Keap1,Kelch 样 ECH 相关蛋白 1;Nrf2,核因子红细胞 2 相关因子 2;OXPHOS,氧化磷酸化;ROS,活性氧;SD,标准差。参考文献:1. 弗里德赖希共济失调研究联盟。什么是 FA?可从 https://www.curefa.org/understanding-fa/what-isfriedreichs-ataxia/ 获取。访问日期:2024 年 11 月。2. Koeppen AH。J Neurol Sci。2011;303(1-2):1-12。3. Campuzano V 等人。Hum Mol Genet。 1997;6(11):1771-1180。 4.Nachun D 等人。哈姆·摩尔·热内特。 2018;27(17):2965-2977。 5.弗里德赖希共济失调研究联盟。 Friedreich 共济失调临床管理指南 (FRDA)。可从 https://frdaguidelines.org/ 获取。访问时间:2024 年 11 月。 6. Campuzano V 等人。科学 。 1996;271(5254):1423-1427。 7.Gatchel JR 等人。纳特·热内特。 2005;6(10):743-755。 8. Bürk K. 小脑共济失调。 2017;4:4。 9.潘道夫·M·尼罗尔·吉内特。 2020;6(3):e415。 10. 汉森 E 等人。世界心脏病杂志。 2019;11(1):1-12。 11.Chiang S 等。神经化学国际公司。 2018;117:35-48。 12. González-Cabo P,帕劳 F. J Neurochem。 2013;126(补编1):53-64。 13. Llorens JV 等。神经科学前沿。 2019;13:75。 14. Petrillo S 等人。国际分子科学杂志。 2017;18(10):2173。 15.D'Oria V 等人。国际分子科学杂志。 2013;14(4):7853–7865。 16. Itoh K 等人,基因发育. 1999;13(1):76-86。17. Santos R 等人,抗氧化还原信号. 2010;13(5):651-690。

弗里德赖希共济失调的大脑结构和退化分期:ENIGMA‐共济失调工作组的磁共振成像体积测量
来自 1 澳大利亚维多利亚州墨尔本莫纳什大学中央临床学院神经科学系;2 澳大利亚维多利亚州克莱顿莫纳什大学莫纳什生物医学成像系;3 澳大利亚维多利亚州克莱顿莫纳什大学心理科学学院特纳大脑与心理健康研究所;4 意大利 Bosisio Parini IRCCS Eugenio Medea 科学研究所神经影像科;5 奥地利因斯布鲁克医科大学神经病学系;6 意大利那不勒斯费德里科二世大学高级生物医学科学系;7 澳大利亚维多利亚州帕克维尔默多克儿童研究所 Bruce Lefroy 中心;8 澳大利亚维多利亚州帕克维尔墨尔本大学;9 德国哈勒 (萨勒) 大学医院放射医学系大学诊所和放射科门诊; 10 德国埃森杜伊斯堡-埃森大学埃森大学医院神经内科;11 意大利博洛尼亚大学电气、电子和信息工程系“Guglielmo Marconi”;12 德国亚琛工业大学神经内科;13 JARA-BRAIN 分子

ataxia -IU印第安纳波利斯学者
钙化纤维 - fa¼评估钙三醇在弗里德里希共济失调患者的神经系统症状中的影响; Chop¼费城儿童医院; DPUFAS¼氘化的多不饱和脂肪酸; ELVIS-FA¼FRDA研究者通过Elamipretide启动了研究(IIS); IMF¼前药富马酸酯(MMF)的前体; IRCCS¼ISTITUTODI RICOVERO E CURA A Carattere Scientife; FDA¼美国食品和药物管理局;男性和女性患者的弗里德里希(Friedreich)共济失调的镜架¼弗里德里希(Friedreich)的共济失调; Frda¼Friedreich共济失调; MFARS¼修饰的Friedreich共济失调评级量表;线粒体线粒体; Friedreich的共济失调患者的Moxie¼RTA408胶囊; NAD¼NAD¼NICINAMIDE腺嘌呤二核苷酸; NDA¼新药申请FDA; NRF¼核呼吸因子; PK/PD¼药代动力学/药物动力学; PPAR¼过氧化物酶体增殖物激活受体; TAT¼转录剂。

2024年11月12日至15日,英国伦敦
本研讨会将提供有关共同的共济失调研究和临床试验环境中使用的常见人类MRI获取方法,分析工具和结果指标的介绍性概述。Edwin Chan(香港中国大学)11:15磷酸二酯酶抑制剂通过纠正果蝇中的Cofilin Pathway和Mitochrial分布来改善Friedreich的共济失调状况Edwin Chan(香港中国大学)11:15磷酸二酯酶抑制剂通过纠正果蝇中的Cofilin Pathway和Mitochrial分布来改善Friedreich的共济失调状况