XiaoMi-AI文件搜索系统
World File Search SystemGATA1

DYRK1A 和 GATA1 在 21 三体巨核细胞生成中的协同作用
简介:患有唐氏综合症 (DS) 或 21 三体综合症 (T21) 的儿童罹患暂时性异常髓系造血 (TAM) 和唐氏综合症急性巨核细胞白血病 (ML-DS) 的风险较高 (1, 2)。TAM 是一种新生儿前白血病,由胎儿时期 T21 与 GATA1s 的独特遗传相互作用引起,GATA1s 是关键造血转录因子 GATA 结合蛋白 1 (GATA1) 的 N 端截短异构体。TAM 和 ML-DS 母细胞均以 GATA1 体细胞突变为特征,从而产生 GATA1s (3, 4),但 ML-DS 母细胞还会获得“第三次打击”突变,通常是在表观遗传调节因子或黏连蛋白复合物成员中 (5, 6)。值得注意的是,在缺乏 T21 的个体中,生殖细胞 GATA1s 突变会导致先天性贫血、血小板减少和/或中性粒细胞减少,但与白血病无关 (7, 8),这证实了 GATA1s 和 T21 共同促进白血病的必要性。细胞周期在造血发育过程中受到精确控制。GATA1 已被证实能抑制细胞周期进程和增殖,并通过阻止转录激活因子 E2Fs 与其下游靶标结合来促进造血细胞的终末分化 (9–11)。Rb/E2F 通路对细胞周期调控至关重要,通常受 GATA1 抑制;然而,由于 GATA1 N 端对这种相互作用至关重要,GATA1s 无法抑制激活因子 E2Fs (9–11)。 GATA1 还抑制 GATA2(GATA 结合蛋白 2),GATA2 是一种造血转录因子,对造血干细胞 (HSC) 和巨核细胞扩增至关重要,在 ML-DS 中经常过表达 (12)。由于没有 N 端结构域,GATA1s 无法正确下调 GATA2,导致 HSC 和巨核细胞过度增殖 (13, 14)。

DYRK1A 和 GATA1 在 21 三体巨核细胞生成中的协同作用
患有唐氏综合征 (DS) 或 21 三体综合征 (T21) 的患者罹患暂时性异常骨髓增生 (TAM) 和急性巨核细胞白血病 (ML-DS) 的风险较高。TAM 和 ML-DS 都需要 GATA1 的产前体细胞突变,从而产生截短的异构体 GATA1。单个 21 号染色体 (HSA21) 基因与 GATA1 协同作用以进行白血病转化的机制很难研究,部分原因是具有野生型 GATA1 (wtGATA1) 或 GATA1 的人类细胞模型有限。HSA21 编码的 DYRK1A 在 ML-DS 中过度表达,可能成为治疗靶点。为了确定 DYRK1A 如何与 GATA1 协同影响造血,我们使用基因编辑破坏了同源 T21 诱导多能干细胞 (iPSC) 中 DYRK1A 的所有 3 个等位基因,这些干细胞具有和不具有 GATA1 突变。出乎意料的是,造血分化表明 DYRK1A 缺失与 GATA1 结合会导致巨核细胞增殖增加和成熟度降低。这种增殖表型与 D 型细胞周期蛋白的上调和 Rb 的过度磷酸化有关,从而允许 E2F 释放并解除其下游靶标的抑制。值得注意的是,DYRK1A 缺失对具有 wtGATA1 的 T21 iPSC 或巨核细胞没有影响。这些令人惊讶的结果表明,DYRK1A 和 GATA1 可能协同抑制 T21 中的巨核细胞增殖,并且 DYRK1A 抑制可能不是 GATA1 相关白血病的治疗选择。

509。骨髓衰竭和癌症易感综合征:先天性:海报II
HSP70伴侣GATA1,关键红细胞转录因子。没有HSP70,GATA1很容易通过caspase3。没有GATA1,关键红系分化靶标的转录未被激活,导致细胞凋亡和红细胞分化的延迟。3先前在RPL突变模型中已经报道了HSP70在DBA中的作用。4

解析唐氏综合征相关白血病模型中巨核细胞生成的逐步突变损伤
突变并可以检查巨核细胞分化和其他疾病表型的渐进性扰动。在本期的 JCI 中,Arkoun 和同事使用分步技术将 GATA1 、 MPL 和 SMC3 突变体引入患有或不患有 DS 的人的诱导多能干细胞 (iPSC) 中实现了这一目标 (9)。研究人员揭示了每种变体的个体贡献以及它们如何与 T21 协同导致 DS-AMKL。作者使用 CRISPR/Cas9 技术进行分步基因编辑,生成了 20 个不同的二体和三体 iPSC 克隆,这些克隆包含 GATA1、MPL W515K 和 SMC3 杂合缺失 (SMC3 +/–) 的组合,并通过功能分析验证了这些变化。 MPL 是血小板生成素的跨膜受体,是巨核细胞成熟为血小板所必需的。胞内结构域通过与 JAK2 相互作用介导信号传导。MPL 515 位点的多个功能获得性氨基酸置换通过血小板生成素依赖性激活 JAK/STAT 通路导致骨髓增生性疾病 (10)。有趣的是,W515K/L 突变也见于 T21 患者的 AMKL 和获得额外 21 号染色体的整倍体个体 (D21) 的白血病中,这可能导致巨核细胞分化改变 (7, 11)。T21 和 Gata1 背景下的 MPL 突变足以诱发小鼠巨核细胞白血病 (12)。此外,作者假设,黏连蛋白基因 SMC3 的单倍体不足通过杂合失活会改变 GATA1 结合的染色质可及性,从而改变巨核细胞分化的转录控制。鉴于这些突变单独导致髓系谱系破坏,逐步 iPSC 模型

Casgevy
背景 Casgevy (exagamglogene autotemcel) 是一种细胞基因疗法,由自体 CD34 + 造血干细胞 (HSC) 组成,通过 CRISPR/Cas9 技术在 BCL11A 基因的红细胞特异性增强子区域进行编辑,以降低红细胞系细胞中的 BCL11A 表达,从而增加胎儿血红蛋白 (HbF) 蛋白质的产生。Casgevy 由患者自身的 HSC 制备而成,这些 HSC 是通过血液分离程序获得的。自体细胞富含 CD34 + 细胞,然后通过电穿孔引入 CRISPR/Cas9 核糖核蛋白 (RNP) 复合物进行体外基因组编辑。RNP 复合物中包含的向导 RNA 使 CRISPR/Cas9 能够在 BCL11A 基因的红细胞特异性增强子区域的关键转录因子结合位点 (GATA1) 处精确地断裂 DNA 双链。编辑的结果是,GATA1 结合被破坏,BCL11A 表达降低。这种减少反过来导致伽马珠蛋白表达增加和下游胎儿血红蛋白形成 (1)。Casgevy 输注后,编辑后的 CD34 + 细胞植入骨髓并分化为 BCL11A 表达降低的红细胞谱系细胞。BCL11A 表达降低导致红细胞中 γ 珠蛋白表达和 HbF 蛋白产生增加。在患有严重镰状细胞病的患者中,HbF 表达可降低细胞内血红蛋白 S (HbS) 浓度,防止红细胞镰状化并解决疾病的根本原因,从而消除血管闭塞性危象 (VOC)。在患有输血依赖性 β-地中海贫血的患者中,γ-珠蛋白的产生可改善 α-珠蛋白与非 α-珠蛋白的不平衡,从而减少无效红细胞生成和溶血并增加总血红蛋白
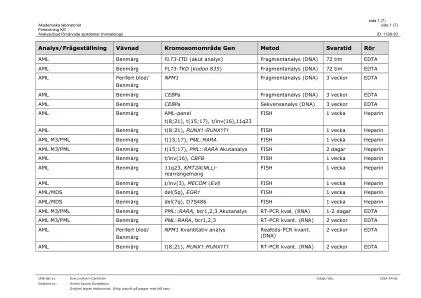
分析提供获得的疾病(血液学)
leukemiutringbenmärg/blod mylood面板*(ABL1,ANKRD26,ASXL1,ATRX,BCOR,BCOR,BCORL1,BRAF,CALR,CALR,CBL,CBL,CBL,CBL,CDKN2A,CDKN2A,CEBPA,CEBPA,CEBPA,CSF3R,CSF3R,CSF3R,CUX1,DDX41,DDX41,DNMT3A fbxw7, FLT3, GATA1, GATA2, GNAS, HRAS, Idh1, Idh2, Ikzf1, jak2, jak3, kdm6a, kit, kraas, kmt2a, mpl, myd88, NF1, Notch1 (INKLUSIVE 3´UTR), NPM1, NRAS, PDGFRA, PHF6, PPM1D, Pten, Ptpn11, Rad21, Runx1, Samd9, SAMDL9, Setbp1, SF3B1, SMC1A, SMC3, SRSF2, Stag2, Stat3, Stat5B, Tet2, TP53, U2AF1, WT1, ZRSR2, BTK, plcg2, terc) Div>

RPS19 编辑的 Diamond-Blackfan 贫血模型...
简介戴蒙德-布莱克凡贫血 (DBA) 是一种罕见的先天性骨髓衰竭疾病,通常在婴儿期表现为大细胞性贫血和红细胞减少症 (1, 2)。DBA 与腭裂、肾脏和心脏缺陷、生长迟缓等身体异常以及某些癌症风险增加有关 (3, 4)。虽然发育不全性贫血是儿童的主要特征,但老年患者也可能出现骨髓细胞减少、全血细胞减少和免疫缺陷,表明造血干细胞 (HSC) 受损 (5, 6)。经典的 DBA 是由 20 个小亚基或大亚基核糖体蛋白 (RP) 基因中的 1 个发生种系杂合功能丧失突变引起的,导致核糖体的生物合成和/或功能缺陷。较不常见的是,GATA1 (7)、EPO (8)、ADA2 (9) 和 TSR2 (10) 的突变会导致 DBA 样增生性贫血。最常见的 DBA 基因是 RPS19,大约 25% 的患者检测到突变。接下来最常见的突变基因是 RPL5 (~7%)、RPS26 (~7%) 和 RPL11 (~5%) (1)。目前对 DBA 的治疗方法包括铁螯合慢性红细胞输注;糖皮质激素(可促进红系祖细胞扩增)和异基因造血干细胞移植 (HSCT),所有这些疗法都与严重毒性有关。DBA 相关红系衰竭的机制尚不完全清楚。对患者造血干细胞和祖细胞 (HSPC) 的分析显示,红系祖细胞扩增存在缺陷,并伴有红系祖细胞病理性凋亡 (1, 11–14)。可能的解释包括整体翻译受损 (15, 16);BAG1 (17)、CSDE1 (17) 和 GATA1 (18, 19) 等红细胞生成所必需的转录本的选择性翻译受损;由于

RPS19 编辑的 Diamond-Blackfan 贫血模型...
简介戴蒙德-布莱克凡贫血 (DBA) 是一种罕见的先天性骨髓衰竭疾病,通常在婴儿期表现为大细胞性贫血和红细胞减少症 (1, 2)。DBA 与腭裂、肾脏和心脏缺陷、生长迟缓等身体异常以及某些癌症风险增加有关 (3, 4)。虽然发育不全性贫血是儿童的主要特征,但老年患者也可能出现骨髓细胞减少、全血细胞减少和免疫缺陷,这表明造血干细胞 (HSC) 受损 (5, 6)。经典的 DBA 是由 20 个小亚基或大亚基核糖体蛋白 (RP) 基因中的 1 个发生种系杂合功能丧失突变引起的,导致核糖体的生物合成和/或功能缺陷。较不常见的是,GATA1 (7)、EPO (8)、ADA2 (9) 和 TSR2 (10) 的突变会导致 DBA 样增生性贫血。最常见的 DBA 基因是 RPS19,大约 25% 的患者检测到突变。接下来最常见的突变基因是 RPL5 (~7%)、RPS26 (~7%) 和 RPL11 (~5%) (1)。目前对 DBA 的治疗方法包括铁螯合慢性红细胞输注;糖皮质激素(可促进红系祖细胞扩增)和异基因造血干细胞移植 (HSCT),所有这些疗法都与严重毒性有关。DBA 相关红系衰竭的机制尚不完全清楚。对患者造血干细胞和祖细胞 (HSPC) 的分析显示,红系祖细胞扩增存在缺陷,并伴有红系祖细胞病理性凋亡 (1, 11–14)。可能的解释包括整体翻译受损 (15, 16);BAG1 (17)、CSDE1 (17) 和 GATA1 (18, 19) 等红细胞生成所必需的转录本的选择性翻译受损;由于

用于斑马鱼组织特异性基因破坏的 CRISPR/Cas9 载体系统
CRISPR/Cas9 基因组编辑技术极大地促进了多种生物体内和体外基因的靶向失活。在斑马鱼中,只需将向导 RNA (gRNA) 和 Cas9 mRNA 注射到单细胞阶段胚胎中,即可快速生成敲除系。在这里,我们报告了一种简单且可扩展的基于 CRISPR 的载体系统,用于斑马鱼的组织特异性基因失活。作为原理证明,我们使用带有 gata1 启动子的载体来驱动 Cas9 表达,以沉默与血红素生物合成有关的 urod 基因,特别是在红细胞谱系中。Urod 靶向在斑马鱼胚胎中产生了红色荧光红细胞,重现了在 yquem 突变体中观察到的表型。虽然 F0 胚胎表现出嵌合基因破坏,但这种表型在稳定的 F1 鱼中似乎非常明显。该载体系统构成了空间控制基因敲除的独特工具,大大拓宽了斑马鱼功能丧失研究的范围。

AD 2 - LCRA - 1 - 1 英国军用 AIP 阿克罗蒂里
1. 注意。10/28 号跑道的混凝土末端和距离 10 号跑道入口 2000 英尺以内的跑道区域在潮湿时容易打滑,特别是如果发现雨后有积水。 2. 注意。鸟击风险高,特别是在春秋两季的迁徙季节。 3. 电缆 插图:a. 10 号跑道 - 373 米/1223 英尺。b. 28 号跑道 - 374 米/1227 英尺。正常操作 - 两条电缆均已拆除。快速喷气机操作 - App 电缆已拆除,超限电缆已升起。 4. 电路 a. 方向。10 号跑道 RHC:28 号跑道 LHC。b. 高度。(i)正常 - 1100 QNH。(ii)重度 - 1600 QNH。 (iii) FJ 低空转弯 - 600 QNH。5. TKOF 后保持 Rwy Tr 直到飞过海面。6. 附加频率:Talkdown 240·05、GATA1 240·1、GATA2 355·0、APS Ops 369·45、SAR 252·8。