XiaoMi-AI文件搜索系统
World File Search SystemGPX4

肿瘤特异性 GPX4 降解增强铁死亡......
脂质过氧化依赖性铁死亡已成为一种新兴的肿瘤治疗策略。然而,目前的策略不仅选择性地诱导恶性细胞中的铁死亡,而且还同时触发免疫细胞中的铁死亡,这可能会损害抗肿瘤免疫力。在这里,我们使用 In-Cell Western 检测结合无偏药物筛选,确定化合物 N6F11 是一种铁死亡诱导剂,可触发谷胱甘肽过氧化物酶 4 (GPX4)(一种关键的铁死亡抑制剂)的降解,特别是在癌细胞中。N6F11 不会导致免疫细胞(包括树突状细胞、T 细胞、自然杀伤细胞和中性粒细胞)中的 GPX4 降解。从机制上讲,N6F11 与癌细胞中 E3 泛素连接酶三联基序 25 (TRIM25) 的 RING 结构域结合,从而触发 TRIM25 介导的 K48 连接 GPX4 泛素化,导致其蛋白酶体降解。从功能上讲,N6F11 治疗导致铁死亡癌细胞死亡,从而启动由 CD8 + T 细胞介导的 HMGB1 依赖性抗肿瘤免疫。N6F11 还增强了晚期癌症模型中针对 CD274/PD-L1 的免疫检查点阻断,包括由 KRAS 和 TP53 突变驱动的胰腺癌基因工程小鼠模型。这些发现可能建立一种安全有效的策略来增强铁死亡驱动的抗肿瘤免疫。

mtor-sirt1,nrf2,gpx4 ferroptosis
此外,还需要进行大规模验证研究以确认新兴分子和技术干预的功效和安全性[45]。纳入现实世界数据和多功能分析的强大临床试验对于建立这些创新方法的临床实用性至关重要,并确保它们成功地转化为常规的患者护理。此外,跨越分子生物学,生物工程和数据科学等多学科专业知识的整合对于有效实施这些综合方法至关重要[46]。在使用先进技术(例如AI,数字双胞胎和机器人生物技术)的使用的道德和监管方面也将是这些变革性解决方案的未来发展和部署的关键方面。

自闭症儿童早期脑过度生长的细胞机制:GPX4 水平升高和对铁死亡的抵抗
患有不成比例的巨脑症 (ASD-DM) 的自闭症患者,其脑部相对于身高较大,智力障碍的发生率高于脑部大小正常的自闭症儿童,面临的认知挑战也比患有平均脑容量的自闭症儿童更严重。这种神经表型背后的细胞和分子机制仍不甚明了。为了研究这些机制,我们从正常发育的非自闭症儿童和患有和不患有不成比例的巨脑症的自闭症儿童中产生了人类诱导性多能干细胞。我们利用磁共振成像和全面的认知和医学评估对这些儿童进行了纵向评估,从 2 岁到 12 岁。我们发现,来自 ASD-DM 儿童的神经祖细胞 (NPC) 表现出更高的细胞存活率和抑制的细胞死亡,同时伴有

o r i g i n a l r e s a r c h氯胺酮通过靶向lncrna pvt1/mir-214-3p/gpx4
背景:肝癌在全球范围内排名前四名,需要有效且安全的治疗。铁凋亡是由铁依赖性脂质过氧化驱动的一种新型的调节细胞死亡形式,被认为是癌症的有前途的治疗靶标。在这项工作中,我们旨在研究麻醉氯胺酮对肝癌的增殖和铁毒性的影响。方法:通过细胞计数套件8(CCK-8),菌落形成和5-乙基-2'-脱氧尿苷(EDU)分析检测到细胞活力和增殖。铁凋亡是由Fe 2+,脂质活性氧(ROS)和丙二醛(MDA)的水平确定的。通过实时PCR测定法检查了LNCPVT1,miR-214-3p和谷胱甘肽过氧化物酶4(GPX4)的RNA水平。临床肝肿瘤样品,以检测长期非编码RNA LNCPVT1,miR-214-3p和GPX4的水平,并通过Pearson比较测试评估它们的相关性。进行了荧光素酶报告基因测定和RNA下拉,以确定LNCPVT1,miR-214-3p和GPX4 3ʹUTR之间的结合。结果:氯胺酮在体外和体内显着抑制了肝癌细胞的生存力和增殖,以及刺激的铁毒性,以及LNCPVT1和GPX4的表达降低。LNCPVT1直接与miR-214-3p相互作用,以阻碍其作为GPX4海绵的作用。LNCPVT1的耗竭加速了活癌细胞的铁凋亡,而miR-214-3p抑制和GPX4过表达却逆转了这种作用。MiR-214-3p抑制和GPX4过表达也抑制了氯胺酮诱导的细胞生长抑制和铁凋亡。结论:在这项工作中,我们确定氯胺酮抑制了肝癌细胞的生存能力并诱导了铁毒性,并确定了LNCPVT1/ MIR-214-3P/ GPX4轴的可能调节机制。关键字:肝癌,氯胺酮,LNCPVT1,mir-214-3p,GPX4

trim32通过增强K63- ...
铁凋亡被认为是脊髓损伤(SCI)激活的细胞死亡途径之一。然而,管理此过程的确切调节机制仍然鲜为人知。在这里,这项研究确定了TRIM32,一种E3泛素连接酶,是神经元铁毒性神经元的关键增强子。trim32通过加速GPX4的降解来促进神经元萎缩,这是甲状腺毒性的必不可少的抑制剂。神经元中TRIM32的条件缺失显着抑制神经元的铁肿瘤并促进神经元存活,最终改善了SCI后小鼠运动功能恢复。然而,TRIM32的过表达表现出严重的神经元丧失和行为功能差,可能会因抑制剂liproxstatin-1而减弱。从机械上讲,TRIM32与GPX4相互作用,在K107处促进了GPX4的K63连接的泛素化修饰,从而增强了GPX4的p62依赖性自噬降解。此外,ROS-ATM-CHK2信号通路在S55处磷酸化的TRIM32,进一步导致SCI后GPX4泛素化和降解以及随后的神经元肥胖病,表明ROS和TRIM32之间的阳性反馈回路循环循环。在临床上,SCI患者可显着促进脂质过氧化。这些发现表明,TRIM32是一种神经元螺氏凋亡增强剂,在SCI后通过促进K63连接的泛素化和随后的p62依赖性自载体脱离GPX4的GPX4,对小鼠的神经元存活和运动型恢复有害。

引用:Sha J,Liu W,Wu J,Yanping W,Li X,Ren H,Pang Z,Zhang W,Lee CS,WangP。
Figure 5 (Color online) (a) CLSM images of HepG-2 cells after incubation with TBPCP and TBCP (5 μM) under hypoxic conditions and two-photon irradiation (940 nm, 50 mW, 2 min) followed by staining with C11-BODIPY 581/591, Hoechst 33342, and Fer-1.对于C11-Bodipy 581/591:λEX= 561 nm; λEM= 570–620 nm。用于氧化的C11-Bodipy 581/591,λEX= 488 nm,λem= 500–530 nm。比例尺:10μm。(b)在深色和白光照射下用TBPCP和TBCP(5μM)处理的HEPG-2细胞的GSH水平(400-700 nm,200 mW/cm 2,10 min)。(c)在不同TBPCP处理的条件下,HEPG-2细胞中GPX4表达和GPX4的相对表达的蛋白质印迹分析。(d)在不同TBCP处理的条件下,HEPG-2细胞中GPX4表达和GPX4的相对表达的蛋白质印迹分析。误差线代表平均值±SD(每组n = 3), * p <0.05,** p <0.01,*** p <0.001。(e)TBPCP和TBCP处理的线粒体和核形态的生物-TEM(5μM)HEPG-2细胞在不同的处理后,比例尺:500 nm:500 nm。
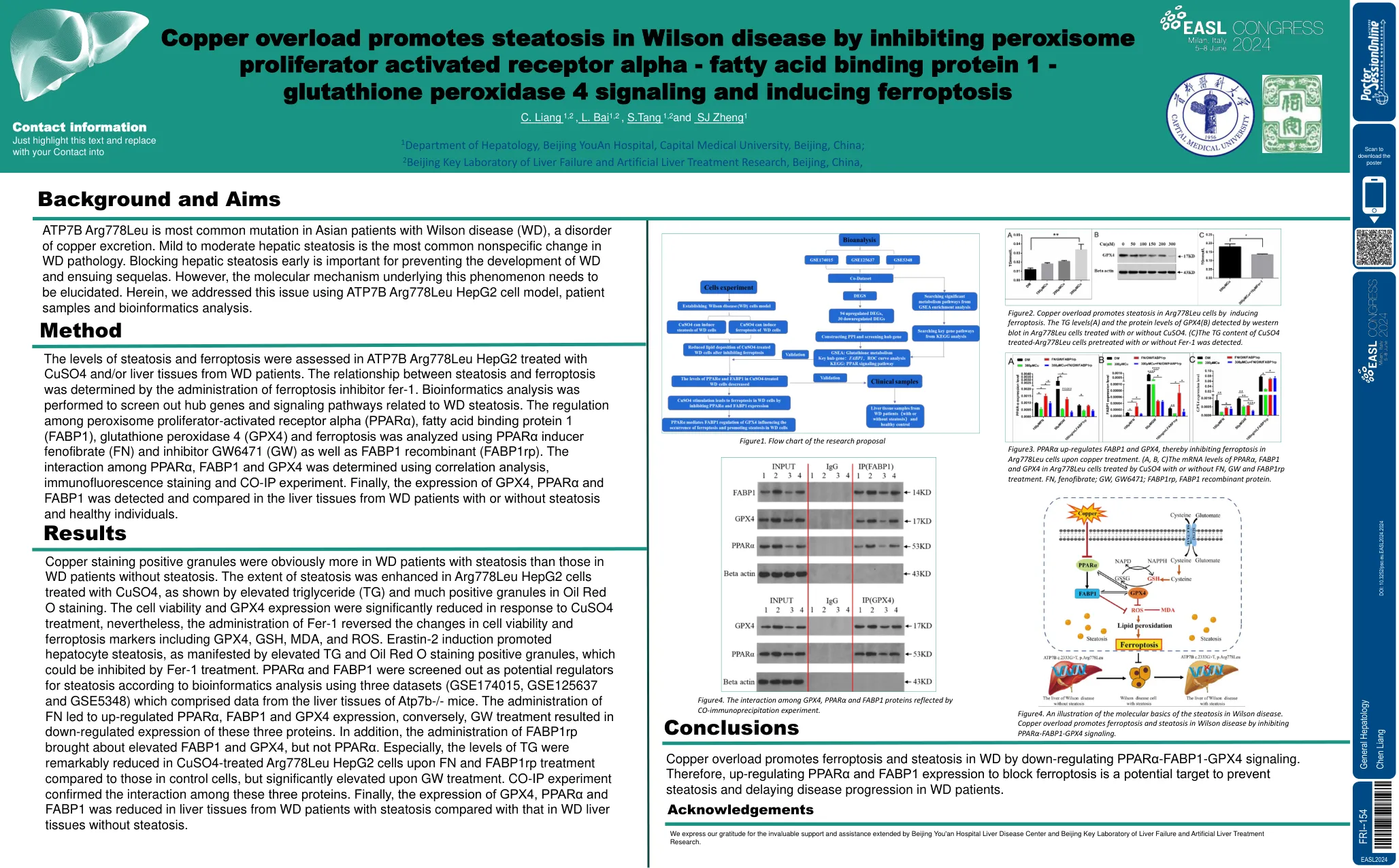
铜超载通过抑制过氧化物酶体促进威尔逊疾病中的脂肪变性
铜染色阳性颗粒显然比没有脂肪变性的WD患者的脂肪变性患者更多。在用CUSO4处理的ARG778LEU HEPG2细胞中,脂肪变性的程度得到了增强,如甘油三酸酯升高(TG)和油红色O染色中的阳性颗粒所示。对CUSO4处理的响应,细胞活力和GPX4表达显着降低,但是,FER-1的给药逆转了细胞生存能力和包括GPX4,GSH,GSH,MDA和ROS在内的细胞生存力和铁凋亡标记的变化。erastin-2诱导促进了肝细胞脂肪变性,如TG和油红O染色阳性颗粒所表现出的,这可以受到FER-1处理的抑制。PPARα和FABP1作为脂肪变性的潜在调节剂进行筛选,该数据集包含来自ATP7B-小鼠的肝脏组织的数据。FN的施用导致了上调的PPARα,FabP1和GPX4表达,相反,GW处理导致这三种蛋白的表达下调。此外,FABP1RP的给药带来了FABP1和GPX4的升高,但没有PPARα。,与对照细胞中的FN和FABP1RP处理后,经过FN和FABP1RP处理后CUSO4处理的ARG778LEU HEPG2细胞的TG水平明显降低,但在GW处理后却显着升高。co-IP实验证实了这三种蛋白质之间的相互作用。最后,与没有脂肪变性的WD肝组织相比,WD患者的肝组织中GPX4,PPARα和FABP1的表达降低。

Sorafenib sensitizes melanoma cells to vemurafenib through ferroptosis
Ferroptosis is a recently recognized form of regulated cell death driven by small molecules or conditions that induce lipid-based reactive oxygen species (ROS) accumulation, and this iron-dependent form of cell death is morphologically and genetically distinct from apoptosis, necroptosis, and autophagy (8,9). Ferroptosis is characterized by cell volume shrinkage and increased mitochondrial membranes and is mediated by iron-dependent lipid peroxide accumulation (10). The ferroptosis-inducing compounds, such as erastin and Ras selective lethal 3 (RSL3) could inactivate cellular glutathione (GSH)-dependent antioxidant defenses, leading to the accumulation of toxic lipid ROS (11,12). Glutathione peroxidase 4 (GPX4) is a key antioxidant enzyme that is responsible for removing lipid hydroperoxides within biological membranes (8). Once GPX4 inactivation, GSH will loses ability in removing the local peroxidase reaction, which eventually lead to a lipid ROS accumulation and ferroptosis.

分子机制研究,抑制与少年败血症相关的小鼠的GPX4介导的肌吞噬作用的Angong Niuhuang药丸的粗提取物
抽象背景:败血症相关的脑病(SAE)是与败血症相关的器官功能障碍的一种普遍形式。没有伴随的明显的中枢神经系统(CNS)感染,但它具有死亡率的重大风险,可能导致持久的神经系统并发症。Angong niuhuang药丸(AGNH)在诸如脑缺血,脑部创伤和败血症等疾病中的功效已经建立了良好。尽管如此,AGNH在SAE进展中的特定调节作用和基本机制仍未探索。方法:脂多糖(LPS)处理用于构建SAE大鼠模型。Berderson的神经检查评分系统用于评分。通过酶联免疫吸附测定(ELISA)或相应的商业试剂盒检查基因和铁含量的水平。通过自动凝血分析仪确认了凝血酶原时间(PT),激活的部分血栓质蛋白时间(APTT),凝血酶时间(TT)和纤维蛋白原(FIB)水平。通过苏木精(HE)染色评估了神经元的数量和形态。蛋白质表达是通过蛋白质印迹确定的。结果:在AGNH或Deatecamine(DFO,铁毒性抑制剂)治疗后,LPS治疗介导的伯德森从未通过LPS治疗介导的功能评分增加,这表明AGNH改善了少年SAE小鼠的神经行为功能。此外,AGNH改善了年轻SAE小鼠的炎症和凝结参数。AGNH促进了少年SAE小鼠的神经元生长和减轻神经元损伤。此外,AGNH抑制了年轻SAE小鼠的氧化应激。最后,证明AGNH促进了与核因子2相关因子2(NRF2)/谷胱甘肽过氧化物酶4(GPX4)信号传导途径,通过上调NRF2和GPX4蛋白表达式。结论:这项研究表明,通过调节NRF2/GPX4信号通路,AGNH具有抑制GPX4诱导的少年SAE小鼠纤维毒性的能力。这一突破意味着AGNH作为SAE的治疗剂有前途的前景。

新型的甲基转移酶G9a抑制剂通过NRF2/HO-1途径诱导多发性骨髓瘤的铁毒性
癌症生长[17]。 我们怀疑MM细胞中DCG066诱导的凋亡模式与甲状腺毒作用有关,因此我们预先处理了ARH-77和RPMI-8226细胞具有氧化肌毒化抑制剂(FER-1)的RPMI-8226细胞,并通过添加DCG0666666666的诱导剂,并通过添加了MOSTBIDEBSBIDEBSBIDED。 我们发现,与单独的MM细胞中的DCG066处理组相比,Erastin和Fer-1能够很好地逆转和促进DCG066诱导的凋亡(图癌症生长[17]。我们怀疑MM细胞中DCG066诱导的凋亡模式与甲状腺毒作用有关,因此我们预先处理了ARH-77和RPMI-8226细胞具有氧化肌毒化抑制剂(FER-1)的RPMI-8226细胞,并通过添加DCG0666666666的诱导剂,并通过添加了MOSTBIDEBSBIDEBSBIDED。我们发现,与单独的MM细胞中的DCG066处理组相比,Erastin和Fer-1能够很好地逆转和促进DCG066诱导的凋亡(图4a,p <0.001)。随后,用不同浓度(0,3 µm,5 µm,8 µm)的DCG066对ARH-77和RPMI-8226细胞进行处理,效力诱变的主要调节剂的蛋白质水平(GPX4和SCL7A11)(GPX4和SCL7A11)分析了GPX11的蛋白质水平。 DCG066浓度(图4b)。因此,我们假设DCG066导致MM