XiaoMi-AI文件搜索系统
World File Search SystemH3K4

H3K4三甲基化调节癌症免疫
摘要 随着肿瘤免疫调控和免疫治疗的进展,组蛋白修饰在建立抗肿瘤免疫能力中的作用不断被发现,开发表观遗传药物(epi-drugs)与免疫检查点抑制剂或嵌合抗原受体-T细胞疗法的联合疗法有望提高免疫治疗的效益。组蛋白H3赖氨酸4三甲基化(H3K4me3)是肿瘤免疫调控中一个关键的表观遗传修饰,深度参与调节肿瘤免疫原性、重塑肿瘤免疫微环境、调节免疫细胞功能。但如何整合这些理论基础,创造新的基于H3K4三甲基化的治疗策略并优化现有疗法仍不清楚。本综述中,我们阐述了H3K4me3及其修饰物调控抗肿瘤免疫的机制,并探索了H3K4me3相关药物与免疫疗法联合治疗的潜力。了解 H3K4me3 在癌症免疫中的作用将有助于开发新的表观遗传疗法和推进基于免疫疗法的联合方案。

KDM1A 维持转录增强子的全基因组稳态
转录增强子能够对后生动物的基因表达进行精确的时空控制。组蛋白 H3 赖氨酸 4 (H3K4me1) 的单甲基化富集是转录增强子的主要染色质特征。赖氨酸 (K) 特异性脱甲基酶 1A (KDM1A,也称为 LSD1) 是一种 H3K4me2/me1 脱甲基酶,可在小鼠胚胎干细胞 (mESC) 分化过程中使干细胞增强子失活。然而,其在未分化 mESC 中的作用仍不清楚。在这里,我们表明 KDM1A 在未分化和谱系定向细胞中都积极维持最佳增强子状态。KDM1A 占据了未分化 mESC 中的大部分增强子。增强子处的 KDM1A 水平与其底物 H3K4me2、H3K27ac 和增强子处的转录呈现明显的正相关性。在缺乏 Kdm1a 的 mESC 中,这些增强子中的大部分获得了额外的 H3K4 甲基化,同时伴有 H3K27 乙酰化增加以及增强子 RNA (eRNA) 和靶基因表达增加。在有丝分裂后的神经元中,KDM1A 的缺失会导致神经元活动依赖性增强子和基因的过早激活。总之,这些结果表明 KDM1A 是一种多功能的增强子调节器,并充当变阻器,通过平衡增强子处的 H3K4 甲基化来维持最佳增强子活性。
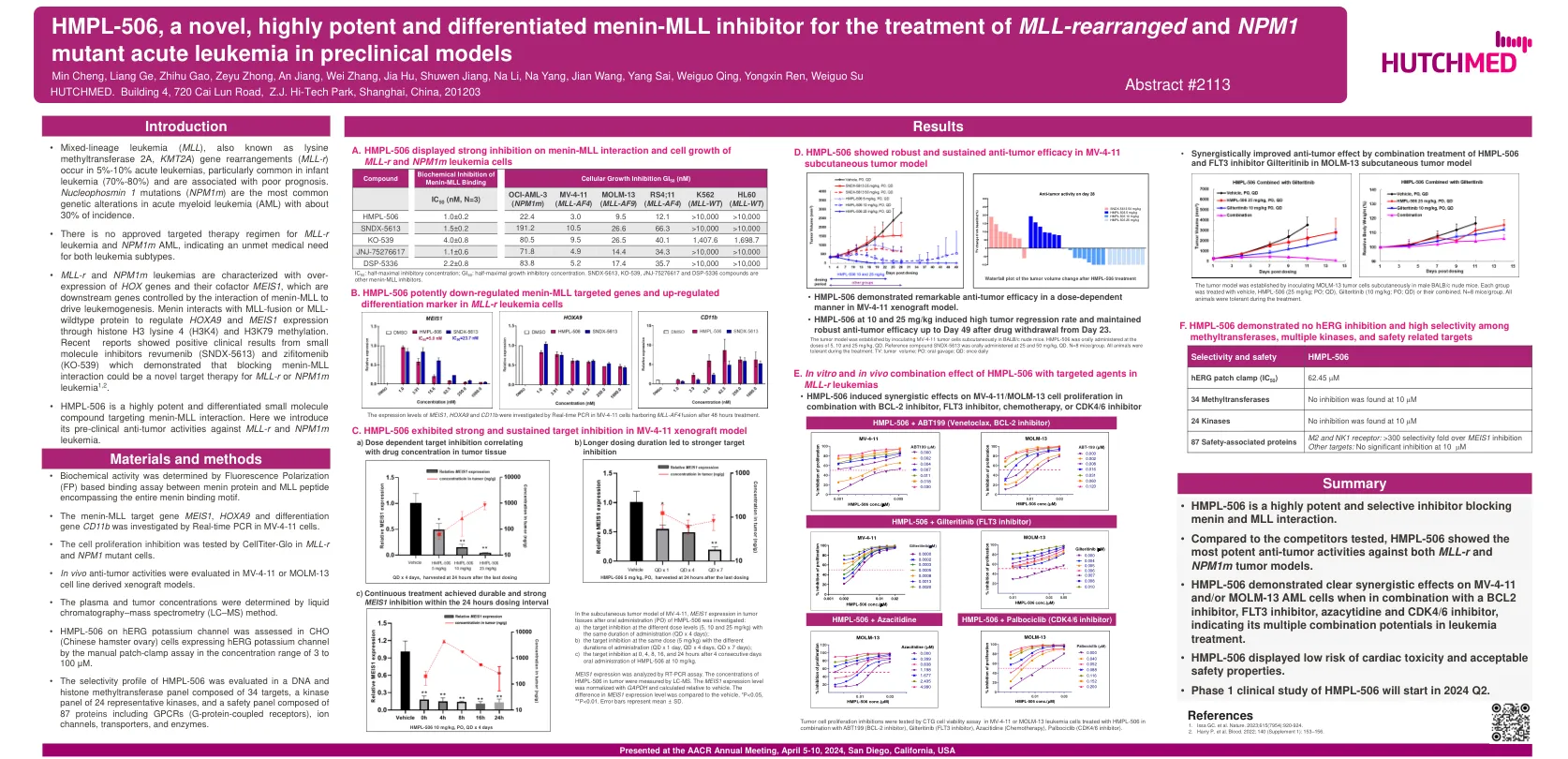
HMPL-506,一部小说,高度有效的和分化的Menin ...
HOX基因及其辅助因子Meis1的表达,它们是由Menin-Mll相互作用以驱动白血病发生的下游基因。Menin与MLL融合或MLL-野生型蛋白相互作用,通过组蛋白H3赖氨酸4(H3K4)和H3K79甲基化来调节HOXA9和MEIS1表达。最近的报道显示,小分子抑制剂rebumenib(SNDX-5613)和Zifitomenib(KO-539)的阳性临床结果,这些结果表明,阻断Menin-MLL相互作用可能是对MLL-R-R或NPM1M1M1M1M1M1M1M1M1M1,2的新型靶标治疗。

IL-9由白血病干细胞分泌的诱导Th1- ...
摘要:在急性髓样白血病(AML)中,白血病和祖细胞(LSC和LPC)与骨髓(BM)微环境中的各种细胞类型相互作用,调节其扩张和分化。为了研究BM与LSC和LPC在BM中CD4+和CD8+ T细胞的相互作用,我们通过公正的高通量相关网络分析分析了它们的转录组和预测细胞细胞相互作用。我们发现,AML患者BM中的CD4+ T细胞被激活并倾斜到Th1极化,而IL-9产生(TH9)CD4+ T细胞不存在。与正常的造血干细胞(HSC),LSCS产生的IL-9和相关模型相反,在LSC中预测IL9是激活AML中CD4+ T细胞的主要轮毂基因。功能验证表明,CD4+ T细胞中的IL-9R信号传导导致JAK-STAT途径的激活,从而诱导KMT2A的上调,KMT2C,KMT2C创造物,导致在裂解酶4(H3K4)对组蛋白H3上的甲基化(H3K4)上的甲基化,以促进经典的访问性和转录率激活。这种诱导的Th1扭转,增殖和效应子细胞因子分泌,包括干扰素(IFN) - ɣ和肿瘤坏死因子(TNF)-α。 IFN-ɣ,较小的扩展由活化的CD4+ T细胞产生的TNF-α诱导LSC的膨胀。根据我们的发现,LSC中的高IL9表达和BM渗透CD4+ T细胞中高IL9R,TNF和IFNG表达与AML的总体存活率较差有关。因此,由AML LSC分泌的IL-9塑造了Th1链的免疫环境,该环境通过分泌IFN-ɣ和TNF-α来促进其扩张。

二价染色质作为癌症治疗靶点
肿瘤细胞异质性是有效设计靶向抗癌疗法的主要障碍。药物治疗前表型不同的肿瘤细胞亚群的多样化分布容易导致反应不一致,导致敏感癌细胞被消除,而耐药亚群却不受伤害。很少有人提出量化与个体癌细胞异质性相关的变异性并将其对临床结果的不良影响降至最低的策略。在这里,我们报告了一种计算方法,该方法可以合理设计涉及针对染色质修饰剂的表观遗传药物的组合疗法。我们制定了一个二价转录因子的随机模型,使我们能够表征三种不同的定性行为,即:双稳态、高基因表达和低基因表达。分析结果与实验数据的比较确定了所谓的双稳态和高基因表达行为可以分别与未分化和分化细胞类型识别。由于具有异常自我更新潜能的未分化细胞可能表现出癌症/转移起始表型,我们在双稳态子集合内的异质性背景下分析了表观遗传药物组合的效率。虽然单靶向方法大多无法规避肿瘤异质性所代表的治疗问题,但组合策略的效果要好得多。具体而言,预计更成功的组合涉及组蛋白 H3K4 和 H3K27 去甲基化酶 KDM5 和 KDM6A/UTX 的调节剂。然而,那些涉及 H3K4 和 H3K27 甲基转移酶 MLL2 和 EZH2 的策略预计效果较差。我们的理论框架为开发一种计算机模拟平台提供了连贯的基础,该平台能够识别最适合治疗管理异质癌细胞群非均匀反应的表观遗传药物组合。
414 科学文摘
背景:在训练免疫过程中,单核细胞和巨噬细胞经历功能和转录重编程以达到激活状态,这是由启动刺激诱导的,并导致对后续触发的反应性增强。类风湿性关节炎 (RA) 患者的单核细胞表现出与训练免疫表型一致的特征。瓜氨酸化蛋白质如瓜氨酸化波形蛋白 (c-波形蛋白),在 RA 中起损伤相关模式的作用,可能与训练免疫过程有关。目的:我们旨在研究 c-波形蛋白是否在健康个体中体外诱导训练免疫。方法:通过 Ficoll-paque 离心和使用 CD3/CD19/CD56 磁珠进行负选择,从健康供体的外周血 (EDTA 血液,n=22;白膜,n=6) 中分离单核细胞。用 c-波形蛋白 (0.1 μg/ml) 刺激细胞 24 小时,5 天后用大肠杆菌脂多糖 (LPS) (10 ng/ml) 再次刺激。用 ELISA 测定第 6 天细胞培养上清液中的蛋白质和乳酸释放量。应用 RT-PCR 和/或 Western Blotting 测量 mRNA 和/或蛋白质表达。使用配体受体糖基捕获技术 LRC-TRi-CEPS 识别 c-波形蛋白的候选细胞表面靶点。通过染色质免疫沉淀检查组蛋白 H3 在赖氨酸 4 (H3K4) 处的甲基化。结果:用瓜氨酸化波形蛋白进行启动可诱导人类单核细胞进行训练,这可通过用 LPS 重新刺激后分泌的白细胞介素 6 (IL-6) 水平显著增加来证明(增加 1.29 倍,n=22,p<0.001)。同样,趋化因子 CXCL1 和 CCL20/巨噬细胞炎症蛋白 3a 的释放也显著增加(分别增加 1.81 倍和 2.32 倍,n=14,p 值均<0.001)。LRC-TRiCEPS 能够识别配体 c-波形蛋白的 STING 细胞表面受体。事实上,c-波形蛋白通过磷酸化诱导与 STING 信号通路有关的 TBK1 的激活,而用共价小分子 H151 (2μM) 抑制 STING 可消除这种影响。此外,H151 通过减少 IL-6 释放和表达来抑制训练免疫(分别减少 1.61 倍和 1.93 倍,n=5)。训练的单核细胞也表现出高乳酸产生(经引发与未引发的细胞,n=9,p=0.004),反映了代谢的转变和糖酵解的增加。通过抑制 2-脱氧葡萄糖(11mM)的糖酵解代谢途径,可以抵消训练免疫的诱导(IL-6 释放减少 5.32 倍,n=7,p=0.016)。最后,c-波形蛋白诱导 H3K4 甲基化,IL-6 基因启动子中该标记的水平增加。通过使用甲基硫腺苷 (1mM) 来调节表观遗传酶的功能,甲基硫腺苷 (1mM) 可特异性抑制组蛋白甲基转移酶,从而逆转训练后的免疫力(IL-6 释放减少 8.43 倍,n=6,p=0.031)。结论:瓜氨酸化波形蛋白可能通过 STING 和 TBK1 依赖性激活诱导单核细胞的表观遗传修饰和代谢变化,从而导致再刺激后细胞因子和趋化因子产生增强。抑制 STING 信号通路可能是 RA 中髓系激活的新治疗靶点。利益披露:未声明 DOI:10.1136/annrheumdis-2021-eular.3302

新发现的调节癌症支持基因表达的因素
我们预测,只有在两种蛋白质结合时,就会存在一个独特的分子,并且从使用分裂 - 涡轮注释3进行的分析中,我们还发现,许多转录调节剂与该复合物结合起作用。从以上结果来看,已经揭示了BOD1L与setD1a结合,并且比作为DNA修复调节剂更有帮助癌症生长和生存的转录启动子。 ■研究人员的评论(Chiba University医学研究生院Hoshii副教授)我们很高兴能够解决蛋白质 - 蛋白质相互作用的奥秘,这些蛋白质相互作用已经很长时间了。 SETD1A本身也引起了人们的关注,作为儿童疾病和精神分裂症的原因,因此我们希望这一发现将有助于癌症以外的其他疾病的治疗。 ■词汇表注释1)CRISPR平铺方法:一种通过设计基因编辑技术CRISPR/CAS9中用于单个基因的无数SGRNA来全面检查和识别在蛋白质上具有功能的位置的方法。注2)DEPMAP数据库:一个数据库,旨在鼓励发现癌症治疗靶标和开发治疗方法,以1,000多个癌细胞系进行的大规模CRISPR-CAS9筛选的结果。注3)分裂 - 涡轮增压:一种接近依赖性的标记方法,允许识别其周围蛋白质的标记,仅限于两种蛋白质相互作用时。 ■Paper information Paper title: BOD1L mediates chromatin binding and non-canonical function of H3K4 methyltransferase SETD1A Author: Takayuki Hoshii*, Sota Kikuchi, Tomoya Kujirai, Takeshi Masuda, Tomoko Ito, Satoshi Yasuda, Makoto Matsumoto, Bahityar Rahmutulla, Masaki Fukuyo,Takeshi Murata,Hitoshi Kurumizaka,Atsushi Kaneda *负责作者杂志名称:核酸研究doi:10.1093/nar/gkae605■参考材料1纸张1个纸张标题:SetD1A的非静脉功能调节setd1a的非催化功能。 10.1016/j.cell.2018.01.032■参考材料2纸张标题:setD1a在白血病杂志中调节血红素生物合成基因的转录暂停释放杂志名称:细胞报告DOI:10.1016/j.cellep.2022.1111727