XiaoMi-AI文件搜索系统
World File Search SystemHadfield
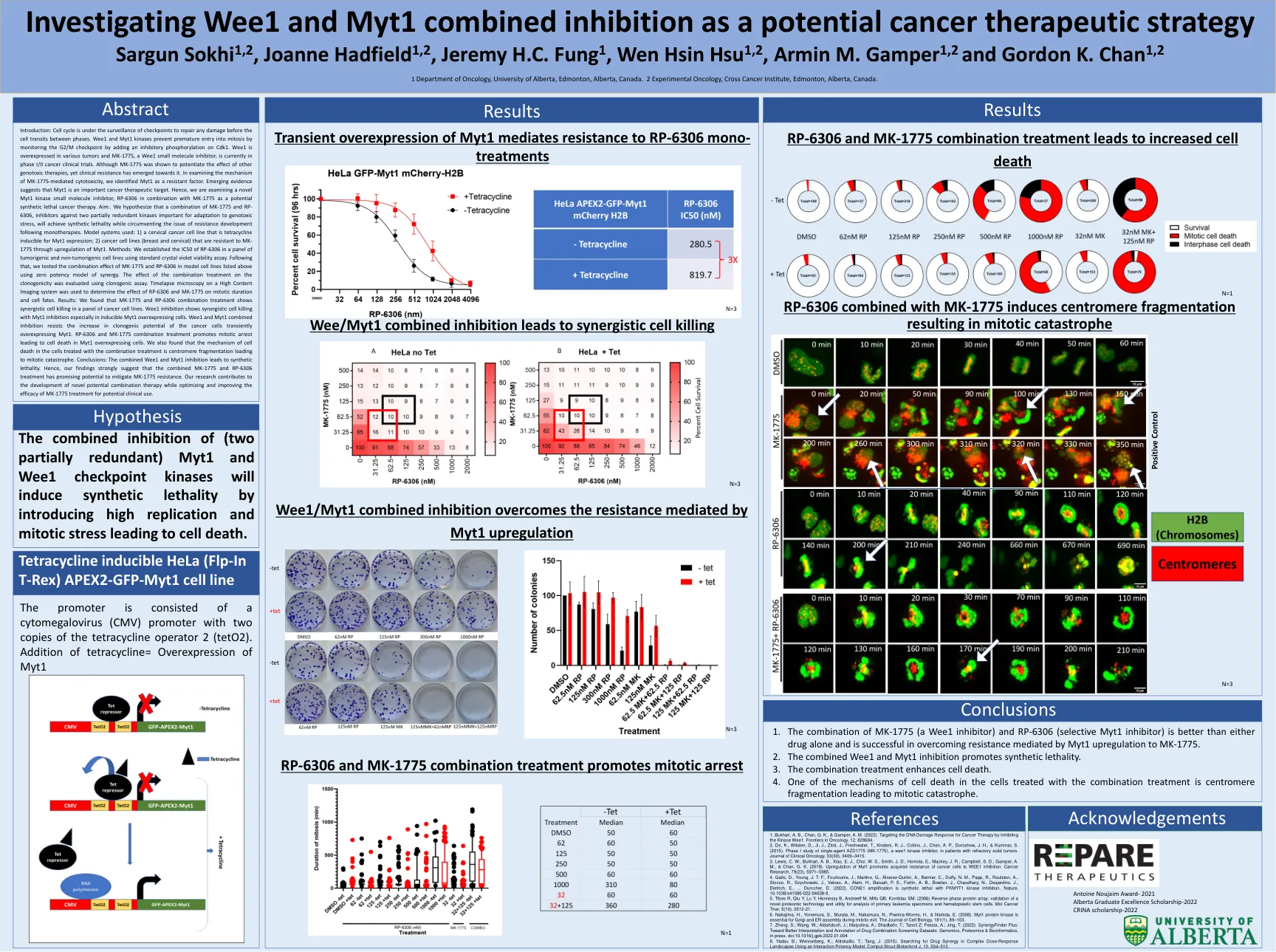
研究WEE1和MYT1作为潜在的癌症治疗策略Sargun Sokhi 1,2,Joanne Hadfield 1,2,Jeremy H.C.真菌1
简介:细胞周期处于检查点的监视下,以修复各个相之间的细胞过渡之前的任何损坏。WEE1和MYT1激酶通过在CDK1上添加抑制性磷酸化来监测G2/M检查点,以防止过早进入有丝分裂。WEE1在各种肿瘤中过表达,而MK-1775(WEE1小分子抑制剂)目前正在I/II期癌症临床试验中。尽管显示了MK-1775可以增强其他遗传毒性疗法的作用,但临床抗性却朝向其抗性。在检查MK-1775介导的细胞毒性的机理时,我们将MYT1确定为抗性因子。新兴证据表明,MYT1是重要的癌症治疗靶点。因此,我们正在研究一种新型的MYT1激酶小分子抑制剂RP-6306与MK-1775结合使用,作为潜在的合成致死性癌症治疗。aim:。我们假设MK-1775和RP-6306的组合是针对两个部分冗余激酶对适应遗传毒性应激很重要的抑制剂,将实现合成的致死性,同时绕过单一层次的耐药性发育问题。使用的模型系统:1)宫颈癌细胞系,是四环素可诱导的MyT1表达; 2)通过上调myt1,对MK-1775具有抗性的癌细胞系(乳房和颈椎)。方法:我们使用标准的晶体紫罗兰色生存力测定法在一组肿瘤和非肿瘤细胞系中建立了RP-6306的IC50。之后,我们使用协同效力模型在上面列出的模型细胞系中测试了MK-1775和RP-6306的组合效应。使用克隆生成测定法评估了组合处理对克隆原性的影响。对高含量成像系统上的时间段显微镜用于确定RP-6306和MK-1775对有丝分裂持续时间和细胞命运的影响。结果:我们发现MK-1775和RP-6306组合处理表明,癌细胞系中的协同细胞杀死。WEE1抑制作用显示了通过MYT1抑制的协同细胞杀死,尤其是在诱导的MYT1过表达细胞中。 WEE1和MYT1结合抑制作用可抵抗癌细胞瞬时过表达MYT1的克隆发育潜力的增加。 RP-6306和MK-1775组合治疗促进有丝分裂停滞,导致MYT1过表达细胞的细胞死亡。 我们还发现,用联合处理处理的细胞中细胞死亡的机理是丝粒碎片化导致有丝分裂灾难。 结论:合并的WEE1和MYT1抑制导致合成致死性。 因此,我们的发现强烈表明,MK-1775和RP-6306的组合治疗具有减轻MK-1775耐药性的潜力。 我们的研究有助于开发新型的潜在组合疗法,同时优化和提高MK-1775治疗的潜在临床用途的功效。WEE1抑制作用显示了通过MYT1抑制的协同细胞杀死,尤其是在诱导的MYT1过表达细胞中。WEE1和MYT1结合抑制作用可抵抗癌细胞瞬时过表达MYT1的克隆发育潜力的增加。RP-6306和MK-1775组合治疗促进有丝分裂停滞,导致MYT1过表达细胞的细胞死亡。我们还发现,用联合处理处理的细胞中细胞死亡的机理是丝粒碎片化导致有丝分裂灾难。结论:合并的WEE1和MYT1抑制导致合成致死性。因此,我们的发现强烈表明,MK-1775和RP-6306的组合治疗具有减轻MK-1775耐药性的潜力。我们的研究有助于开发新型的潜在组合疗法,同时优化和提高MK-1775治疗的潜在临床用途的功效。

Dada,A。C.,Kaniewski,J.,Gawith,C.,Lavery,M.,Hadfield,R.H.,Faccio,D。和Clerici,M。(2021)近距离最大的两光子纠缠opti
纠缠仍然是基于通信和信息处理协议(例如量子密钥分布(QKD)[1-3],超密集编码[4]和状态传送[5]的许多新兴量子技术的关键要素。迄今为止,基于引导波和自由空间传输的可见和电信波长的启用这些协议的主力是光源[6]。近年来,卫星到地面链接已成为长距离QKD的最有前途的选择[7-12]。卫星到地面QKD的挑战是在日光下的可操作性有限,因为电信和可见频带的背景过多[13]。因此,迄今为止,大多数示例都依赖于夜间操作,只有少数例外[14]。此外,在日光下,基于纠缠或与设备无关的方法仍有待证明。设备独立的实现是指关于QKD设备的工作方式或它们基于哪种量子系统的方式的假设[15,16]。此外,基于卫星的推动通信网络正在导致QKD的范式转移到与设备无关的实现,这些实现必须同时支持FILBRE和自由空间光学链接。2至2.5 µm光谱区域正迅速成为高度有希望的光学电信带,比传统的电信C波段(1550 nm)具有显着优势,这对于在此波段带中的量子源和测量能力至关重要。例如,已经证明2- µm条带在中空核心光子带隙(HCF)[17]中具有最小的损失,这是由于其超低的非纤维性而导致的一种新兴传输 - 纤维替代方案,并且提供了最低的可用延伸度。使用HCFS [18]证明了2- µm区域中2.5 dB/km的损失[18],其范围可进一步减少,超过0.14 dB/km>的最小衰减效果。

大攻角操纵品质评级量表
我在此提交一篇由 Chris A. Hadfield 撰写的论文,题为“高攻角操控品质评定量表”。我已检查了该论文的最终电子版形式和内容,并建议接受该论文作为部分满足理学硕士学位(主修航空系统)的要求。

过渡边缘传感器:在粒子中启用发现...
•Lita,Adriana E.等。“用于量子应用的超导型单光子和光子数解析检测器的开发。”Lightwave Technology Journal 40.23(2022):7578-7597。•Morozov,Dmitry V.,Alessandro Casaburi和Robert H. Hadfield。“超导光子检测器”。当代物理学62.2(2021):69-91。

量子人工智能和 NASA
Eleanor Riefel M. Sohaib Alam Lucas Brady David Bernal Neira Stephen Cotton Zoe Gonzalez Izquierdo Shon Grabbe Stuart Hadfield Aaron Lott Filip Maciejewski Salvatore Mandr`a Jefrey Marshall Gianni Mossi Jason Saied Nishchay Suri Norman Tubman Davide Venturelli 王志辉 现职实习生

军备学习指南 v1-4.pdf - 谢菲尔德市议会
© 谢菲尔德图书馆档案和信息 2013-2015 (v.1.4) 封面插图 - 从左到右: 萨维尔街上的装饰拱门,庆祝爱德华七世国王和亚历山大王后的皇家访问,由约翰布朗公司赞助,1905 年(谢菲尔德地方研究图书馆:图片谢菲尔德 s03198) 贝壳工人,Cammell Laird and Co. Ltd.,谢菲尔德,1916 年(谢菲尔德地方研究图书馆:图片谢菲尔德 s00540) Hadfield's Ltd 生产的 Heclon 12” 封盖贝壳,约 1910 年(谢菲尔德地方研究图书馆:y04811) 图像可以在未经我们许可的情况下复制用于私人或教育用途,但我们要求包含以下确认“[文档参考编号] 来自谢菲尔德图书馆档案和信息收藏”。如果您希望发布、展示或广播本指南中的任何信息,请联系我们。


Robert W. Thacker 2024夏季实习计划纽约气候... Aishwarya Kumar教授(他/他/他的) b'' 请求从COVID-19疫苗征收医疗豁免
Stony Brook University生态与进化系的专业经验2015年至上教授。2019年至今的研究助理,史密森尼热带研究所,巴拿马共和国2015 - 2021年生态与进化系主席,Stony Brook University。2012-2015阿拉巴马大学伯明翰生物学系教授。2012 - 2013年伯明翰阿拉巴马大学生物学系临时主席。2006-2015科学家,UAB艾滋病研究中心。2006-2012伯明翰阿拉巴马大学生物学系副教授。2000-2006伯明翰阿拉巴马大学生物学系助理教授。1998-2000关岛海洋实验室的博士后研究助理。顾问:Valerie J. Paul博士。 1996-1998夏威夷大学太平洋生物医学研究中心的博士后研究助理。 顾问:Michael G. Hadfield博士。 1995-1996关岛海洋实验室的博士后研究助理。 顾问:Valerie J. Paul博士。 1990-1995博士研究,部门 生物学,密歇根大学安阿伯大学。 顾问:Brian A. Hazlett博士(退休)。顾问:Valerie J. Paul博士。1996-1998夏威夷大学太平洋生物医学研究中心的博士后研究助理。顾问:Michael G. Hadfield博士。1995-1996关岛海洋实验室的博士后研究助理。 顾问:Valerie J. Paul博士。 1990-1995博士研究,部门 生物学,密歇根大学安阿伯大学。 顾问:Brian A. Hazlett博士(退休)。1995-1996关岛海洋实验室的博士后研究助理。顾问:Valerie J. Paul博士。 1990-1995博士研究,部门 生物学,密歇根大学安阿伯大学。 顾问:Brian A. Hazlett博士(退休)。顾问:Valerie J. Paul博士。1990-1995博士研究,部门生物学,密歇根大学安阿伯大学。 顾问:Brian A. Hazlett博士(退休)。生物学,密歇根大学安阿伯大学。顾问:Brian A. Hazlett博士(退休)。

TPS 515:agenT-797(不变的自然杀伤 T 细胞)、botensilimab(Fc 增强的 CTLA-4 抑制剂)和 balstilimab(抗 PD-1)的 II 期研究
1. Wilke H、Muro K、Van Cutsem E 等。雷莫芦单抗联合紫杉醇与安慰剂联合紫杉醇治疗既往接受过治疗的晚期胃腺癌或胃食管连接部腺癌患者(RAINBOW):一项双盲、随机 3 期试验。Lancet Oncol。2014 年 10 月;15(11):1224-35。2. Hadfield MJ、Safran H、Purbhoo MA、Grossman JE、Buell JS、Carneiro BA。利用同种异体不变自然杀伤 T 细胞(iNKT)克服对程序性细胞死亡蛋白 1(PD-1)阻断的耐药性。Oncogene。2024 年 3 月;43(10):758-762。3. Bullock AJ、Schlechter BL、Fakih MG 等。 Botensilimab 联合 balstilimab 治疗复发/难治性微卫星稳定转移性结直肠癌:1 期试验。Nat Med。2024 年 9 月;30(9):2558-2567。