XiaoMi-AI文件搜索系统
World File Search SystemHifi

标准化的术语和PACBIO HIFI测序和RAAV基因治疗载体分析的报告
尽管重组腺相关病毒(RAAV)是基因疗法的主要平台,但缺乏标准化的计算分析方法和通过长阅读测序评估每个帽子的内容的报告。PACBIO高度准确的长阅读HIFI测序可以对AAV基因组进行全面表征,但需要生物信息学专业知识来分析,解释和比较结果。为了满足这一需求并提高对功能性病毒有效载荷的理解,我们的工作组建立了标准化的命名法,并报告了RAAV矢量的长阅读测序数据。工作组建议涵盖与矢量纯度(全长与零散基因组)和污染物(宿主DNA,质粒DNA)鉴定有关的关键质量属性(CQA)。通过推荐的协议,我们对从头制造的数据分析揭示了全部和部分填充的衣壳的特异性以及部分/截断的载体物种的高分辨率表征。最后,我们提供了实施此

将 dsDNA 与 ssDNA Oligo 和 NEBuilder HiFi DNA 组装连接起来,创建 sgRNA-Cas9 表达载体
摘要 本应用说明展示了将 sgRNA 序列插入 9.5 kb 载体进行靶向 DNA 组装的便利性。与必须合成并重新退火两个寡核苷酸的传统克隆方法不同,此新方案提供了一种简单的方法来设计寡核苷酸并将其与所需载体组装。NEBuilder HiFi DNA 组装主混合物比传统方法有了显著的改进,特别是在节省时间、易于使用和成本方面。

将 dsDNA 与 ssDNA Oligo 和 NEBuilder HiFi DNA 组装连接起来,创建 sgRNA-Cas9 表达载体
摘要 本应用说明展示了将 sgRNA 序列插入 9.5 kb 载体进行靶向 DNA 组装的便利性。与必须合成并重新退火两个寡核苷酸的传统克隆方法不同,此新方案提供了一种简单的方法来设计寡核苷酸并将其与所需载体组装。NEBuilder HiFi DNA 组装主混合物比传统方法有了显著的改进,特别是在节省时间、易于使用和成本方面。

产品注意:使用HIFI WGS的Agilent Fem Tuls系统快速,准确的DNA尺寸
安捷伦(Agilent)开发了一个基因组质量数量(GQN)度量,用于敏捷的数据分析软件,以评分基因组DNA质量。Prosize软件根据用户定义的指定尺寸阈值的总测量浓度的比例计算出GQN值从0到10。1建议的阈值和可接受的GQN将取决于应用程序和样本准备工作流程。对于整个基因组测序,PACBIO建议启动基因组DNA在10 kb时至少具有7.0或更高的GQN(GQN10KB≥7.0),而GQN为5.0或更高,为30 Kb(GQN30KB≥5.0)的GQN为5.0或更高。图4显示了使用Agilent基因组DNA 165 Kb试剂盒在FEM脉冲系统上分析的人类唾液样本的示例。由于该样品在10 kb和30 kb的GQN阈值以下,因此建议使用短读取消除剂(SRE)试剂盒处理以耗尽分子量下分子量DNA片段,以在DNA剪切之前提高样品质量。请参阅PACBIO库准备协议,以获取有关基因组DNA质量,GQN指南和尺寸选择选项的其他信息。2

技术说明全血样品的HIFI全基因组测序的高通量DNA剪切
库以相等的摩尔方式合并,并使用具有85 pm加载浓度的单个SMRT®细胞在续集®IIE系统上进行测序。QC和测序结果(图3-4,表2)表明1,400 rpm速度设置产生了最佳的HIFI读取长度轮廓。剪切240秒产生的平均HIFI读取长度为17,703 bp,而剪切480秒的平均读数为16,855 bp的平均读取长度。在240和480秒时,更快的1,800 rpm设置覆盖了DNA,导致平均HIFI读取长度分别为13,184 bp和11,658 bp。通常,当使用FastPrep-96剪切DNA时,较小的工作速度较小的时间将导致较大的平均片段长度。
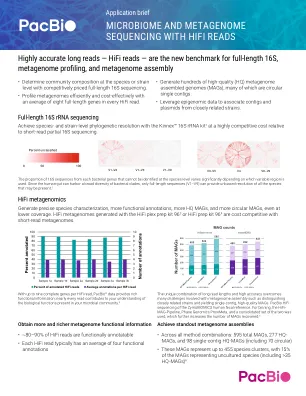
应用简介-使用 HiFi 进行宏基因组测序-...
1. 应用说明 – Kinnex 16S rRNA 试剂盒用于全长 16S 测序 2. Johnson, JS 等人 (2019) 评估 16S rRNA 基因测序在物种和菌株水平微生物组分析中的应用。《自然通讯》。10(1),5029。 3. 程序和清单 – 使用 HiFi plex 制备试剂盒 96 制备多重全基因组和扩增子文库 4. 程序和清单 – 使用 HiFi 制备试剂盒 96 制备全基因组文库 5. Gehrig, J. 等人 (2022) 找到合适的选择:评估短读和长读测序方法以最大限度提高临床微生物组数据的效用。《微生物基因组学》,8(3),10.1099/mgen.0.000794。 6. Portik, DM 等人(2024) 使用长读组装、分箱和合并方法从人类肠道微生物群中高度准确地组装宏基因组。bioRxiv。doi:https://doi.org/10.1101/2024.05.10.593587 7. 概述 – HiFi 应用选项和测序建议。8. 程序和清单 – 使用条形码引物扩增细菌全长 16S 基因。9. 程序和清单 – 从 16S rRNA 扩增子制备 Kinnex 文库

技术说明 - 使用 DNA Genotek Oragene 设备收集并使用 Nanobind 试剂盒提取的唾液 DNA 样本的 HiFi 测序性能
采集了 30 位捐献者的唾液样本,其中 90% 的分析前 DNA 质量 >2 µg。从 27 个样本中提取了 HMW DNA,其中 93% 的产量 >500 ng。提取后,使用 Qubit dsDNA BR 检测试剂盒对 DNA 进行定量,并使用 Femto Pulse 系统(安捷伦科技公司)进行表征。使用 SMRTbell ® 制备试剂盒 3.0 为部分样本制备 HiFi 文库,并使用 SPRQ™ 化学方法在 Revio 系统上进行测序。每个样本都在一个 Revio SMRT 测序池上进行测序。表 1 总结了五个代表性样本的测序数据。这些样本产生了 4.7 到 15.9 µg 的 HMW DNA。HiFi 测序产量为 119 到 133 Gb 的 HiFi 数据,每个基因组的覆盖率为 27 到 40 倍,足以进行全面的 WGS 变异检测。 75% 到 95% 的读数映射到人类参考基因组 (GRCh38)。

评估HIFI长读取测序与整个基因组Bisulfite测序和甲基化史诗般的Beadchip阵列:利用DNA
DNA甲基化是最丰富,最广泛研究的表观遗传修饰之一,在各种生物学过程中起着至关重要的作用,例如发育,癌症,衰老和复杂疾病。在癌症基因组图集(TCGA)等大型队列研究中,Illumina阵列已被广泛用作高通量筛查的经典平台。但是,这种类型的阵列覆盖了人类基因组中的CpG位点的3%。最新一代的DNA测序技术以PACBIO HIFI系统为例,具有产生长序列读数的独特能力,最高为25千碱基。太平洋生物科学(PACBIO)的最新进步致力于提高每碱基准确性和检测DNA修饰的能力。在这项研究中,我们使用DNA甲基化标准评估了PACBIO HIFI测序的性能。由人DNA在CpG部位酶甲基化的DNA标准和未甲基化的人DNA源自HCT116 DKO细胞系。1 ug。样品被测序为约8倍覆盖范围。DNA甲基化数据,并使用PB-CPG-Tools从BAM文件中提取甲基化值。然后,我们比较了从PACBIO HIFI测序获得的结果与由史诗阵列和整个基因组亚硫酸盐测序(WGB)产生的结果。我们发现WGB和PACBIO HIFI天然DNA甲基化调用表现出很高的一致性,表现优于史诗般的阵列,这两种史诗阵列都与甲基化标准和报道的CPG数量一致。使用甲基化的标准样品,HIFI数据报告约有85%的CpG位点的甲基化比大于90%,平均基因组宽93%。同样,WGBS数据显示了约85%的CpG位点的甲基化比大于90%,平均基因组宽95%。相比之下,Epic阵列仅报告40%的CpG位点的甲基化比大于90%,而整个基因组中平均为87%。这些结果表明,HIFI长读取测序可以准确检测到接近100%甲基化的区域的DNA甲基化信号。我们的研究提供了对检测DNA甲基化模式的PACBIO HIFI测序表现的见解及其作为史诗阵列的替代方案的潜力。这项研究的发现说明了如何将DNA甲基化标准用作评估DNA甲基化调用模型的基础真实参考。

Lentipool V2人CRISPR库 - 用户指南实现特定于目标效率-truecut-hifi-cas9-
为了更好地了解Truecut Hifi Cas9的高保真度,我们评估了HEK293基因组中的其他基因。使用TEG-Seq进行了更多全基因组筛查,以检测HEK1,HEK4,VEG1和VEG3基因中的靶标。数据表明,Truecut Hifi Cas9比WT-CAS9和供应商I高保真CAS9蛋白产生的脱离目标较少(图1)。将每个编辑位点的脱靶编辑百分比与靶向编辑的百分比进行了比较,以确定相应站点的脱靶/靶向概率比。每个编辑事件均与其概率比(图1A)绘制,并根据概率将OFF目标的总数分组(图1B)。结果表明,与WT-CAS9和供应商I高保真CAS9相比,Truecut Hifi Cas9产生的脱靶编辑明显少得多。truecut hifi cas9只有一个非目标编辑的概率> 10%。相比之下,WT-CAS9和供应商I高保真CAS9分别具有16和6折叠目标(图1B)。
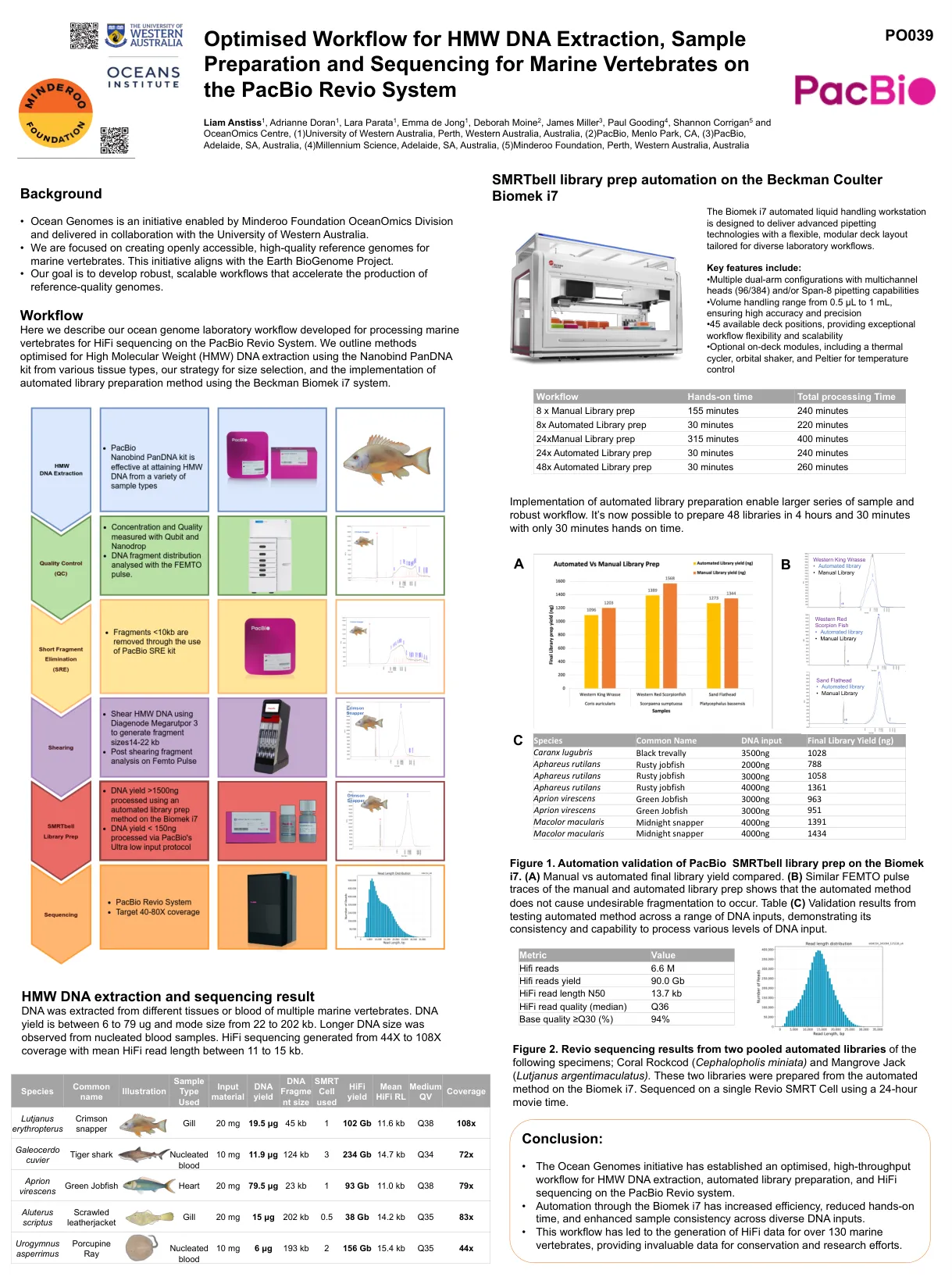
优化的HMW DNA提取,样品制备和测序的工作流程Pacbio Revio System上的海洋脊椎动物
•海洋基因组计划已在PACBIO Revio系统上建立了用于HMW DNA提取,自动化库制备和HIFI测序的优化的高通量工作流程。•通过Biomek i7自动化的效率提高,动手时间降低,并提高了各种DNA输入的样品一致性。•此工作流程导致了130多个海洋脊椎动物的HIFI数据,提供了无价的保护和研究工作的数据。