XiaoMi-AI文件搜索系统
World File Search SystemHuntingtin

2023 年亨廷顿病治疗会议 - 第 1 天
基因编码的蛋白质完全相同。这种情况很少发生,但它表明 DNA 代码中存在一些对亨廷顿舞蹈症很重要的东西。科学家们认为这些 CAA 中断会改变亨廷顿基因通过体细胞不稳定性发生改变的方式,但事实证明并非如此。这意味着还有更多的工作要做
约翰霍普金斯基因组学 DNA 诊断实验室
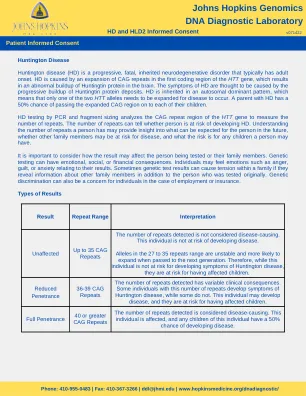
亨廷顿病 (HD) 是一种渐进性、致命性、遗传性神经退行性疾病,通常在成人时期发病。亨廷顿病是由 HTT 基因第一个编码区 CAG 重复扩增引起的,这会导致大脑中亨廷顿蛋白异常积聚。人们认为,亨廷顿病的症状是由亨廷顿蛋白沉积物的渐进性积聚引起的。亨廷顿病以常染色体显性模式遗传,这意味着两个 HTT 等位基因中只需有一个扩增即可发病。患有亨廷顿病的父母有 50% 的机会将扩增的 CAG 区域遗传给他们的每个孩子。通过 PCR 和片段大小测定进行的亨廷顿病检测会分析 HTT 基因的 CAG 重复区域以测量重复次数。重复次数可以判断一个人是否有患亨廷顿病的风险。了解一个人的重复次数可以洞察这个人未来的情况、其他家庭成员是否可能有患病风险以及这个人可能生育的孩子的风险。

简介 亨廷顿氏病 (HD) 是一种遗传性疾病...
简介亨廷顿舞蹈症 (HD) 是一种遗传性疾病,其特征是认知、身体和心理症状逐渐发展。该疾病是由亨廷顿舞蹈症 (HTT) 基因中 CAG 三核苷酸重复序列延长引起的,从而产生了突变型亨廷顿蛋白 (mHTT)。mHTT 在神经元中的积累会导致功能障碍并最终导致细胞死亡[1]。目前,尚无针对亨廷顿舞蹈症的有效治疗方法,治疗重点是控制症状和改善生活质量。然而,基因治疗已成为一种有前途的解决该疾病根本原因的方法。基因治疗在直接改变基因表达或纠正基因缺陷方面已显示出成功,这可能会减缓或阻止亨廷顿舞蹈症的进展。[2]。反义寡核苷酸 (ASO) 或 RNA 干扰 (RNAi) 可以在亨廷顿舞蹈症 (HD) 的基因治疗中实现基因抑制。该方法的主要目的是有效减少突变型HTT mRNA的合成,以建立或抑制致病性突变型亨廷顿蛋白(mHTT)的适当表达。科学家们一直在研究各种基因修饰方法,其中以CRISPR为重点。CRISPR/Cas9可以精确靶向HTT基因中的突变。这可以纠正基因异常或阻止CAG重复片段的扩增。研究表明,使用基因编辑治疗亨廷顿氏病取得了良好的效果。[3,4]

KJELDGAARD 讲座 - Steve Finkbeiner
参考文献:1. Finkbeiner S. 等人 (2004) 包涵体形成降低了突变亨廷顿蛋白的水平和神经元死亡的风险。Nature 431:805–810。2. Finkbeiner S. 等人 (2014) 自噬诱导增强神经元 ALS 模型中的 TDP43 周转和存活率。Nat. Chem. Biol. 10:677–685。3. Finkbeiner S. 等人 (2018) 计算机标记:预测未标记图像中的荧光标记。Cell 173:1–12。4. Finkbeiner S. 等人 (2021b) 使用生物标志物优化的神经网络进行超人细胞死亡检测。Sci. Adv. 7:eabf8142。

罕见疾病
Sci USA )对罕见病的分析显示,期刊文章中关注度最高的是亨廷顿舞蹈症、15,16 重症肌无力、17 ALS、18–20 罕见肿瘤和癌症、21–27 和系统性红斑狼疮 (SLE)。28–30 人们对罕见病的持续科学兴趣意味着研究可以产生高影响力的出版物,例如英国研究人员于 2019 年在新英格兰医学杂志上发表的“针对亨廷顿舞蹈症患者的亨廷顿蛋白表达”31。该文章描述了 Ionis Pharmaceuticals 和 F. Hoffmann–La Roche 设计的寡核苷酸的 I/IIa 期试验结果,该寡核苷酸用于抑制 HTT 的信使核糖核酸 (mRNA),HTT 是导致亨廷顿舞蹈症的主要基因,此后已被引用超过 400 次。

长的体细胞DNA重复扩张驱动亨廷顿疾病的神经退行性
亨廷顿疾病(HD)是一种致命的遗传疾病,其中大多数纹状体投射神经元(SPN)退化。有关HD发病机理的中心生物学问题是亨廷顿蛋白(HTT)基因中引起疾病的DNA重复膨胀(CAG N)如何导致数十年的明显潜伏期后神经变性。遗传的HTT等位基因具有更长的CAG重复急性疾病发作;这种重复的长度也随时间变化,产生了体细胞镶嵌性,调节DNA重复稳定性的基因可能会影响高清年龄。了解细胞的CAG重复长度与其生物学状态之间的关系,我们开发了一种单细胞方法,用于测量CAG重复长度以及全基因组RNA的表达。我们发现,HTT CAG重复在HD-vulnerable SPN中从40-45个CAG扩展到100-500+ CAG,而在其他纹状体细胞类型中则不扩展,而这些长的DNA重复扩展在不同时间通过单个SPN获得。令人惊讶的是,从40个CAGS的体细胞膨胀对基因表达没有明显的影响 - 但是具有150-500+ CAGS的神经元具有深刻的基因表达变化。这些表达的变化涉及数百个基因,并在进一步的CAG重复扩张旁边升级,侵蚀了阳性,然后神经元同一性的负面特征,并在衰老/凋亡基因的表达中达到顶峰。跨高清阶段的纹状体神经元丧失率反映了神经元进入该生物学变形状态的速率。我们得出的结论是,在HD过程中的任何时候,大多数神经元具有无害的(但不稳定的)亨廷顿基因,而HD发病机理几乎是神经元生命的DNA过程。我们的结果表明,纹状体神经元中的HTT CAG重复进行数十年的生物学安静膨胀,因此,由于它们异步越过高阈值,因此SPN会使SPN迅速和异步变性。

HDSA 研究报告
2020 年 10 月,诺华公司宣布他们已获得 FDA 的特殊地位,即孤儿药资格认定,用于研究一种名为 branaplam 的实验性药物对亨廷顿氏病患者的疗效。孤儿药资格认定是一套财务激励措施,使公司开发罕见疾病的治疗方法变得更容易、更有吸引力、更有效。Branaplam 是为治疗儿童遗传性疾病脊髓性肌萎缩症 (SMA) 而开发的。在对动物和人类进行测试时,诺华发现它可以降低亨廷顿氏病的罪魁祸首亨廷顿蛋白的水平。令人兴奋的是:这种药物已知对 SMA 患者是安全的,并且是口服的。下一步将在更多人群中测试 branaplam,以确保它对亨廷顿氏病患者是安全的,并看看它是否有助于缓解亨廷顿氏病症状。诺华计划于 2021 年开始对亨廷顿氏病患者进行 branaplam 试验,获得孤儿药资格将帮助他们实现这一目标。

Aleksandra Kawala-Sterniuk,Dariusz Mikolajewski
亨廷顿氏病(HD)是一种罕见的,无法治愈的神经退行性疾病[31,40,89],以乔治·亨廷顿(George Huntington)的名字命名,他于1872年首次报道了该疾病[5,28,55,59]。最初,该疾病被称为亨廷顿的舞蹈。最初的案例由约翰·埃利奥特森(John Eliotson)在1932年清楚地描述了,有关HD的第一篇论文于1942年由查尔斯·沃特斯(Charles Waters)发表[5]。如上所述,HD是与运动,精神病和认知障碍有关的神经退行性疾病[16,19,26,40,52,66,89]。是由亨廷顿蛋白(HTT)基因中的胞质 - 腺嘌呤 - 瓜氨酸(CAG)三核苷酸重复扩张引起的,该基因产生了突变的亨廷顿蛋白(MHTT)蛋白质,具有异常长的多丁胺重复产生[26,55,69,69,82]。这是一种单基因疾病[59]。大多数症状在成年期间出现[82]。它在临床上以自愿运动受损,舞蹈(非自愿的“舞蹈托动物”运动)的发作,肌张力障碍的混蛋和tick虫以及一些精神病症状(焦虑,冷漠,睡眠问题,抑郁症)[5,26,26,26,28,52,66]。运动症状是HD - 唱片的最具特征性症状,它可能导致写作,饮食,步行和平衡维护方面的问题。唱片可以从偶尔的面部抽搐到大而强大的全身运动。与运动相关的症状可能会受到其他疾病的影响,例如精神恶化(压力和/或焦虑)或身体状况,例如感染[28]。在Manifest HD中均未发生抑郁或焦虑[28]。HD引起进行性纹状体神经元变性[26]。在高清患者中还报告了几个视觉域中的一些障碍,例如视觉对象感知,面部情绪识别,视觉工作记忆或视觉空间处理[66]。在疾病的早期阶段,可以检测到后脑皮质的渐进性结构变化,而额叶和颞大脑区域的影响较小[16]。也有可能注意到高脑白疗法中的突变体亨廷顿表达,这可能表明白质变性在临床症状中的重要作用[52,66]。白质损失被认为与认知障碍更高度相关,这表明其在HD临床表现中的完整性特别重要[52]。通常也受到神经胶去云母和壳质神经de虫的影响(其萎缩[75];此外,还报道了皮质和其他大脑区域的广泛神经元丧失[40,52,66,72]。对这种疾病没有有效的治疗方法,因此生物标志物(在例如MRI图像)不仅有助于诊断,而且可以监测此疾病的进度[19,40,89]。最终,帮助HD患者的唯一方法是仅采用症状疗法,以管理和/或最大程度地减少其身体和/或精神症状[19]。大多数临床症状通常发生在30至50岁之间[20,59,76]。从第一次诊断到死亡的典型生命持续时间在15至20年之间[28,52,59,76]。受肺炎,心血管疾病,跌倒,呼吸系统疾病和自杀造成的伤害,受到最常见HD影响的人(第二大常见的死亡原因)[1,11,28,59,76]。

HYPK控制N- ...
核糖体系的N末端乙酰转移酶A(NATA)乙酰酸盐含量为52%的拟南芥中可溶性蛋白。N末端的这种共同翻译稳定稳定的胞质植物蛋白。进化保守的亨廷顿酵母伴侣K(HYPK)促进了植物学中的NATA活性,但在体外,其N末端螺旋A 1抑制了人NATA活性。为了解剖体内Hypk蛋白结构域的调节功能,我们在遗传学工程师CRISPR-CAS9突变体表达了缺乏所有功能域(HYPK-CR1)的催眠片段(HYPK-CR1)或内部删除的HYPK trunct trunct truncing Helix A 1但仍保留了C-term-Nal ubiquitin-cripied(uba-crain-cripied(uba))。我们发现,Hypk的UBA结构域对于以器官特定方式稳定NATA复合物至关重要。HYPK的N末端,包括Helix A 1,对于从各种氨基酸开始的底物上的NATA活性至关重要。因此,在Hypk n末端内仅删除42个氨基酸会导致植物蛋白质组的实质性不稳定,并且较高的耐受性降低了干旱胁迫。

文章利用 CRISPR-Cas9 技术生成亨廷顿氏病的新同源模型
摘要:亨廷顿氏病 (HD) 是一种致命的神经退行性疾病,由亨廷顿基因 (HTT) 外显子 1 的 CAG 重复扩增引起。尽管亨廷顿氏病具有单基因特性,但其发病机制仍未完全了解,目前尚无有效的治疗方法。基因组工程等新技术的发展为疾病建模领域带来了新机遇,并使得具有相同遗传背景的同源模型的生成成为可能。这些模型对于研究疾病的发病机制和药物筛选非常有价值。本文报告了一系列在 HTT 基因座上具有不同 CAG 重复数的纯合 HEK 293T 细胞系的生成,并展示了它们在测试治疗试剂方面的实用性。此外,利用 CRISPR-Cas9 系统,我们纠正了 HD 人类诱导多能干细胞中的突变并生成了 HTT 基因的敲除,从而为 HD 研究提供了一套全面的同源细胞系。