XiaoMi-AI文件搜索系统
World File Search SystemHuntington



亨廷顿氏病
亨廷顿氏病(HD)被称为亨廷顿舞蹈病,是一种很少发生的神经退行性疾病,具有常染色体式遗传模式,其特征是基底神经节中GABA能神经元的逐渐恶化。其他包括皮质下型痴呆症,行为异常,中年精神病和逐渐无意的浮力运动运动。HD的特征是背纹状体(尾状核和壳核)的萎缩,并在成像形态(例如计算机断层扫描(CT)和磁共振成像(MRI)等成像方式上同时扩展侧心室的额叶角。一项分子研究通过鉴定该疾病的标志放大CAG三重态来验证HD的诊断。目前,无法治愈高清,治疗的重点是提供支持性护理和管理症状。涉及医疗保健专业人员,神经科医生和精神科医生的多学科方法对于综合管理至关重要。药物用于减轻运动症状和管理精神病表现。物理和职业疗法有助于维持功能能力并改善生活质量。遗传咨询和社会心理支持对于患者及其家人至关重要。一个额外的关键目标需要进步更精确,可靠的技术,以及时识别和评估高清。通过早期诊断使及时的干预措施和改善的症状管理成为可能。基于临床和成像发现,我们在一名62岁的女性中提出了HD病例。

亨廷顿英格尔斯工业公司
HII 的大部分业务都与美国政府开展,主要是国防部 (DoD)。作为总承包商、主要分包商、团队成员或合作伙伴,该公司参与了许多高优先级的美国国防计划。通过其 Ingalls 部门,HII 是美国海军两栖攻击舰和远征战舰的建造商、美国海岸警卫队国家安全巡逻舰的唯一建造商,也是建造海军现有阿利伯克级 (DDG 51) 驱逐舰舰队的仅有的两家公司之一。通过其 Newport News 部门,HII 是美国核动力航空母舰的唯一设计者、建造者和加油者,也是目前为美国海军设计和建造核动力潜艇的仅有的两家公司之一。任务技术部门提供广泛的服务和产品,包括 C5ISR 系统和操作;人工智能和机器学习在战场决策中的应用;防御和进攻性网络空间战略和电子战;无人自主系统;实时、虚拟和建设性解决方案;平台现代化;以及关键核行动。

亨廷顿县分区条例
第201条(条例#2020-24)第360条(条例#2020-23)第714条(条例#2020-14)从分区条例中删除,并添加到第715条第715条(条例#2020-21)第901节(条例#2020-21)第901节(条例#2020-15)第912条(第2020-2020-10-2020-120-120-16)。第914节(条例#2020-18)第916条(条例#2020-19)第1050条(条例#2020-20)修订于2021年3月29日,第901节(条例#2021-09)第902节(第902条)第902节(条例#2021-09)(第2021-09节)第903节(第903节)(第2021-2021-2021-2021-09)。 (条例#2021-10)第702条,A,2和3(条例#2021-11)修订于2021年7月19日:第201条(条例#2021-16)第604条(条例#2021-17)第705节(第705节)(i,1)(第2021-19条) #2021-15)第902条(条例#2021-15)第903条(条例#2021-15)第904条(条例#2021-15)第912节(条例#2021-21)第912节(第912条)第912节(条例#2021-22)(第2021-22条)第913条(第913条)(第2021-21-21)第91页(第91-21条)(第91-21页)(第913页)(第913条) (条例#2021-22)第1040条(条例#2021-18)本文件的要求与《印第安纳州亨廷顿县的正式发表法令》有所不同,《法令守则》的规定应规定。

亨廷顿镇 - BESS 暂停期延长
背景 BESS 型设施通常由安装在独立、互连存储单元中的多排可充电电池组成。这些设施通常通过在低使用率期间从当地电网获取剩余能量并将其存储起来,以便在高峰需求期间分配回电网来运行。但是,它们也可以用作可再生能源生产设施(如风能和太阳能发电场)生产的电力的直接存储。无论哪种情况,BESS 都可以确保在电网可能出现部分或全部电压不足(通常称为“电压下降”和“停电”)期间的可靠性,从而稳定当地电网。因此,BESS 的支持者认为,这些设施不仅可以在日常或常规基础上加强当地电网,而且可以在需求特别高或当地电网外部的电力传输被切断的紧急情况下加强当地电网。从土地使用的角度来看,BESS 设施通常被认为是低影响用途。一旦设施建成并投入运营,通常不需要定期配备人员,只需要例行维护。这几乎不会造成交通拥堵,也几乎不需要现场停车。这些设施还可以进行远程监控,从而进一步减少交通、现场人员配备和停车。除了出于安全目的之外,BESS 设施的现场照明也基本上是不必要的。没有员工也意味着 BESS 设施几乎没有用水量,相应地,几乎没有污水。噪音(由冷却风扇产生)通常是与 BESS 设施相关的主要潜在重大规划问题;然而,噪音并不总是一个问题,这取决于项目的规模和配置,并且该行业已经在噪音可能成为问题的情况下实施了噪音缓解方法(即隔音屏障和景观美化)。这些设施的反对者对存在高度易燃物质(例如锂离子电池)以及可能的空气和地下水污染提出了公共安全担忧。从历史上看,对此类威胁的担忧是通过将某些用途从住宅区划出并将其限制在高强度工业区来解决的。然而,对于 BESS 来说,这并不总是可行的,因为这些设施必须通过具有足够容量的变电站连接到当地电网,以适应设施和电网之间的传输。BESS 设施和变电站之间的距离越大,传输效率就越低。因此,在某些情况下,设计可行的 BESS 设施通常需要将设施位于住宅区内或附近。亨廷顿镇于 2020 年 10 月颁布了现行的 BESS 分区法规,该法规载于该镇分区法规第 198-68.3 节 (https://www.lilanduseandzoning.com/wp-content/uploads/sites/128/2023/01/Huntington-1)。pdf)。亨廷顿镇不将 BESS 项目分为不同层级,而其他城镇则可能这样做。相反,面积为两 (2) 英亩或更大且距离住宅区 200 英尺以内的设施需要规划委员会特别许可。现行法规似乎对 BESS 项目相当慷慨,允许它们作为所有轻工业区(I-1 至 I-4)以及一般工业区(I-5)和发电站区(I-6)的主要许可用途。如果 BESS 项目占用的项目场地面积不超过 2%,并且为同一处所内的另一栋建筑或设施提供服务,则允许 BESS 项目在这些区域作为附属用途,并在一般商业区(C-6)获得特别许可。亨廷顿镇的 BESS 法规还包括若干设计要求,影响退缩、高度、场地照明和噪音缓解。还需要批准退役计划。到目前为止,长岛十三 (13) 个城镇中已有四 (4) 个采用了 BESS 分区规定:亨廷顿镇、布鲁克黑文镇、南安普敦镇和艾斯利普镇。然而,据悉,目前萨福克县近一半的城镇正在暂停 BESS 设施建设

患有亨廷顿氏病个体的吞咽困难
在该基因中[3,9,10]。在没有HD的个体中,CAG重复的数量通常不超过34。但是,对于那些高清的人,这个数字可以超过40 [9]。CAG重复的数量增加会触发Huntingtin蛋白的产生,从而导致随后的神经元丧失[9]。值得注意的是,神经元丧失在基底神经节中,特别是在尾状核和壳核中,尽管在大脑皮层中也可以观察到它[2]。HD会导致运动,认知和精神疾病[11]该疾病的最具特征性特征是Chorea,这是一种涉及类似舞蹈,非自愿,快速和非疾病型高激动运动的运动障碍[12]。其他运动障碍包括肌肉僵硬(刚度),肌张力障碍(非自愿和长时间肌肉收缩)和运动缓慢(Bradykinesia)[9,13,14]。与该疾病相关的认知和精神疾病包括痴呆,抑郁,人格变化和注意力不足[12]。随着疾病的发展,采用基因检测来确认临床症状,包括运动,认知和行为障碍的混合,引起怀疑[8,13]。虽然唱片是一种常见的初始经验,但随着疾病的发展,肌张力障碍和僵化也表现出来[13,14]。运动功能中的这些运动中断可能会导致吞咽困难(吞咽困难)和过度运动质心等问题[15,16]。除了导致吞咽困难的运动挑战之外,认知问题也会影响吞咽[15]。值得注意的是,抽吸是HD中的主要死亡原因[18]。吞咽困难又可能导致营养不良,脱水和抽吸肺炎[17]。此外,吞咽困难可以促进社会隔离,活动和参与的局限性以及整体生活质量的下降
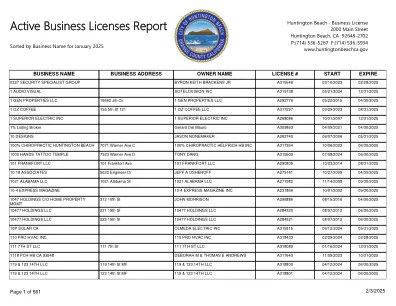
有效营业执照报告 - 亨廷顿海滩
360 运动脊柱与健康公司 18800 Delaware St 1000 360 运动脊柱与健康公司 A299456 06/13/2017 05/31/2025

亨廷顿氏病:研究带来希望
有些人会出现与舞蹈症相关的动作,如行走困难,这会增加跌倒的可能性。有些亨廷顿氏病患者不会出现舞蹈症;相反,他们可能会变得僵硬,很少或根本不动,这种情况称为运动不能。其他人可能一开始患有舞蹈症,但随着病情的发展变得僵硬。除了舞蹈症,有些人还会出现不寻常的固定姿势,称为肌张力障碍。这两种运动障碍可以混合或交替出现。其他症状可能包括震颤(无意识的来回有节奏的肌肉运动)和异常的眼球运动,这些症状通常发生在疾病进展的早期。

亨廷顿镇高级部门高级倡导者......
阿尔茨海默病是一种渐进性疾病,会破坏记忆力和其他重要的心理功能。脑细胞连接和细胞本身会退化和死亡,最终破坏记忆力和其他重要的心理功能。记忆力丧失和思维混乱是主要症状。目前尚无治愈方法,但药物和管理策略可能会暂时改善症状。痴呆症是一组以至少两种大脑功能受损为特征的疾病,例如记忆力丧失和判断力下降。症状包括健忘、社交能力有限和思维能力受损,以至于干扰日常功能。药物和疗法可能有助于控制症状。有些原因是可逆的。长岛阿尔茨海默病协会 300 Broadhollow Road, Suite LL 100, Melville, NY 11747 | (631) 629-6950 https://www.alz.org/longisland