XiaoMi-AI文件搜索系统
World File Search SystemIDUA

神经前体中迁移缺陷的演示
摘要:背景:I型I型Hurler(MPS1-H)是由于IDUA基因的功能丧失突变而导致的严重遗传溶酶体储存障碍。随后的α -iduronidase酶的完全缺乏率直接导致溶酶体中糖胺聚糖(GAG)的进行性积累,从而影响许多组织的功能。因此,MPS1的特征是系统性症状(多器官功能障碍),包括呼吸道和心脏功能障碍,骨骼异常和早期致命神经变性。方法:为了了解MPS1神经病理学的基础机制,我们从两个IDUA等位基因的MPS1-H患者中产生了诱导的多能干细胞(IPSC)。为了避免因IPSC的不同遗传背景而导致的可变性,我们通过通过慢虫方法挽救IDUA表达来建立了IPENIC Control IPSC线。结果:在神经差异后观察到MPS1 -H和IDUA校正的同基因对照之间的明显差异。刮擦测定法显示了MPS1-H细胞的强迁移缺陷。此外,IDUA缺乏对基因表达的影响很大(FDR <0.05的340个基因)。结论:我们的结果表明,迄今为止,溶酶体降解,基因表达和神经运动之间的联系尚不清楚,这可能至少部分解释了MPS1-H患者的表型。

粘多糖化类型的遗传变异
常染色体隐性粘膜性糖尿病I(MPS-I)是一种天生的代谢误差,其中硫酸乙酰肝素和硫酸乙酰肝素硫酸盐由于酶α-iduronidase(IDUA)的缺乏而在细胞中积聚在细胞中,这在直系群中更为普遍。以前,据报道α-辅助酶(IDUA)基因中的变体引起MPS-1表型。本研究的目的是确定十个无关的MPS-1的IDUA基因中的遗传变异,影响了巴基斯坦伊斯兰堡的巴基斯坦医学科学研究所(PIMS),巴基斯坦伊斯兰堡的儿童医院。收集了受影响和未受影响的家庭成员的血液样本,并进行了IDUA基因的测序。在对所有鉴定出的引起疾病变体的硅分析中进行了检查,以检查其对蛋白质结构和功能的影响。对所有MPS-1患者的临床检查均表现出粗糙的面部特征,骨骼畸形,疝气,角膜阴影,腹部延伸和肝肾上腺全球。iDUA基因的测序显示了十种错义变化和八个同义变化。在包括突变品尝器,筛分,多形和普罗普兰在内的有机工具中提出了三种变体,是引起疾病的三种变体。在疾病引起的变异中,在我们的分析家庭的80%中鉴定出了先前报道的错义变体,即c.1469t> c引起p.leu490pro。此外,这是一种新颖的14个核苷酸缺失,即C.568_581DEL AACGTCTCCATGAC引起P.ASN190HIFFS*204和单个核苷酸缺失,即C.784DELC引起P.His262thrfs*55造成了与P.His262thrfs*55造成了与MMS-spy sectize seectize。这项研究报告了80%的筛查家庭中的先前报道的错义变体,一种小说(C.568_581DEL AACGTCTCCATGAC)和先前报道的引起疾病的缺失。

尿酸1-津糖的合成为α-二尿苷酶,β-葡萄糖醛酸酶和乙糖果酶的推定抑制剂**
二芳二酸(L -IDOA)残基硫酸乙酰乙酰胺(HS)和硫酸真皮(DS)中的残基。在MPS I中,低水平的溶酶体IDUA活性会导致HS和DS积聚在细胞中,从而导致包括大脑在内的多个组织和器官的进行性疾病。更严重的MP形式我通常会在生命的前十年内导致智力低下和过早死亡。有两种可用的MPS I:I)使用重组人IDUA静脉注射的酶替代疗法,[2]和II)造血干细胞移植以从健康移植细胞中产生IDUA,但是,两者都有实质性的限制。例如,替代酶不能越过血脑屏障(BBB),因此对神经系统症状没有影响,而造血干细胞移植具有很大的发病率和死亡风险。此外,两种治疗方法都非常昂贵。因此,需要越过BBB并缓解MPS I的神经系统症状的小分子药物的发展是可取的。小分子抑制剂目前正在探索作为溶酶体储存疾病的治疗方法。例如,与累积底物生物合成有关的酶的抑制作用已用于底物还原疗法。最近,研究了有机固核药物Ebselen(2-苯基1,2-苯甲甲硅烷二唑-3(2 h)-One),作为MPS I的潜在底物还原治疗。[3] Ebselen通过抑制L -IDOA生物合成降低了MPS I细胞中的糖胺聚糖积聚。但是,它无法减少MPS I鼠标模型中的糖胺聚糖积累。治疗溶酶体储存疾病的另一种常见小分子方法是药理学伴侣治疗(PCT)。在PCT中,伴侣分子通常是活性位点定向抑制剂,可以结合和稳定突变酶以防止其降解并改善运输到溶酶体。[4]一次在溶酶体的低pH环境中,伴侣分离导致

酶替代疗法粘多糖果
- MPS I型是由基因IDUA突变引起的常染色体隐性溶酶体储存障碍。它的特征是由于酶α-L-核苷酸酶的活性不足或活性不足,其溶酶体积累了硫酸乙酰肝素和皮肤硫酸盐的溶酶体积累。有三种主要的疾病变异:hurler,最严重的早期发作和神经认知能力回归,Hurler-Scheie,中间发作和严重性的形式,以及Scheie,Scheie是最温和的亚型。I型I型患者的体征,症状和严重程度差异很大。最常见的症状包括更严重的形式,脊髓脊髓压缩,角膜云,开态性倍增增生和瓣膜性增生和不足,腕骨型,腕管,腕管,腕管,短隧道和弱点/稳定性。- 美国医学遗传学学院2011年指南通过血清测定法证实了I型I型国家MPS I型,显示出α-L-二维罗苷酶活性的降低。一旦显示患者的酶活性降低,应进行基因检测,该测试应显示IDUA基因突变。两项测试必须证明疾病以确认诊断。- 酶替代(ERT)和干细胞移植(SCT)是MPS类型I的唯一治疗选择。Aldurazyme是MPS I型的唯一FDA批准的酶替代疗法。它被批准为I型MPS的成人和小儿患者以及患有中度至重度症状的Scheie形式的患者。ert应该根据治疗医师的判断开始,并可以在温和的疾病中持续到更重要的临床情况。如果发现患者患有严重疾病或有神经认知能力下降的风险,则应评估他们的SCT。

使用CRISPR/CAS9系统对I型粘多糖含量的潜在脱靶位点的硅胶分析:对人群特异性治疗的影响
粘多糖含量I型(MPS I)是由IDUA基因编码的α-l-二维罗苷酶缺乏引起的。用CRISPR/CAS9的治疗正在开发用于治疗,但是必须对脱靶效应进行详细研究。本研究旨在评估旨在纠正MPS I患者最常见变体的SGRNA的可能脱离目标(P.TRP402 *)。在至少一个人群中,在这些序列中鉴定了共272个势靶序列,并在这些序列中鉴定出84个聚形态位点,其频率等于或大于1%。在大多数情况下,多态性位点减少了脱靶裂解的机会,并创建了新的PAM,这表明了这种分析的重要性。这项研究强调了使用I型粘二糖化病在人群特定环境中筛选靶点的重要性,作为与所有治疗治疗有关的问题的一个例子。我们的结果可以为已经在临床上使用的其他目标提供更广泛的应用,因为它们可能会影响CRISPR/CAS9的安全性和效率。
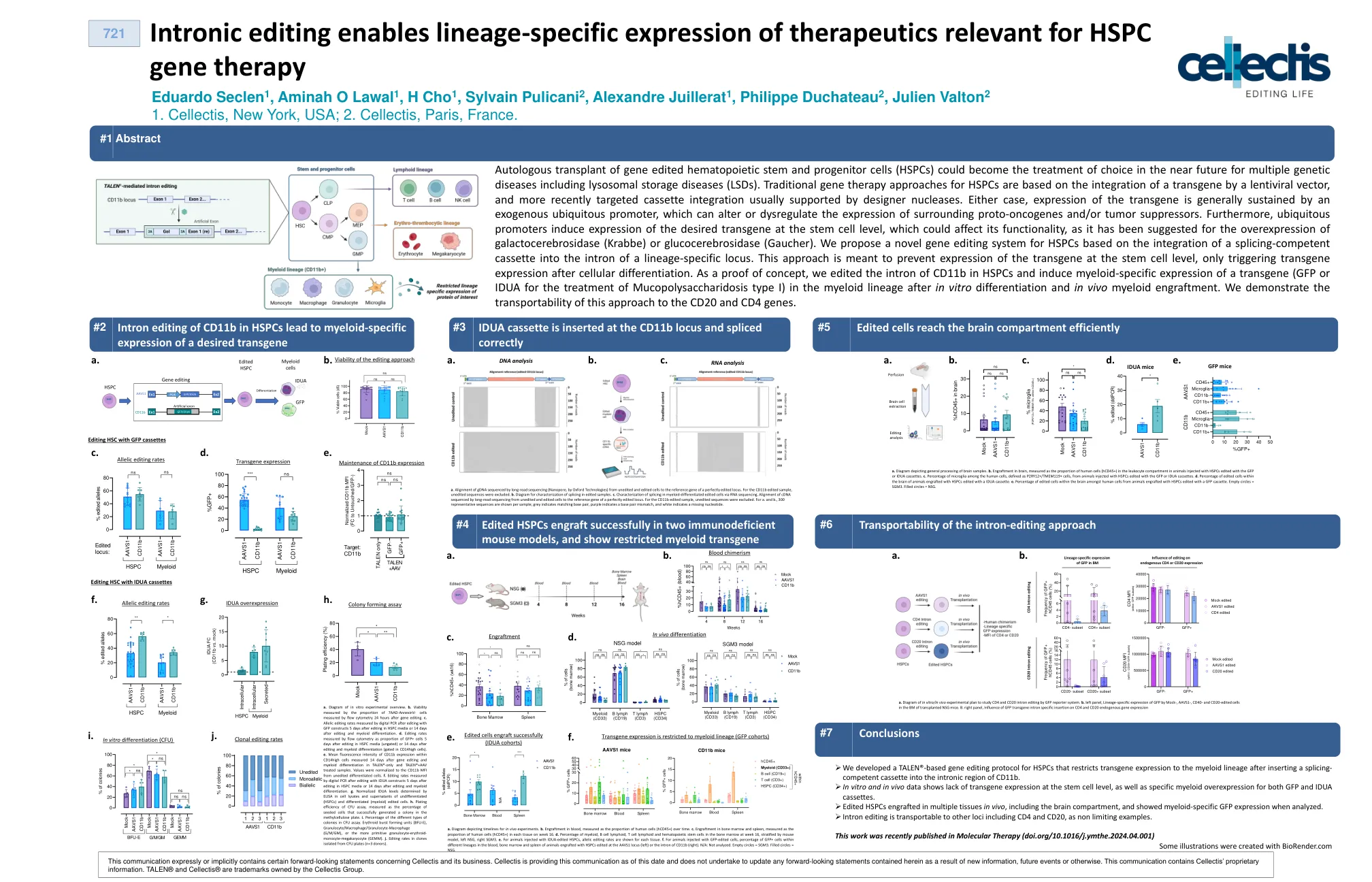
内含子编辑可以使谱系特异性表达与HSPC基因治疗相关的治疗剂
基因编辑的造血茎和祖细胞(HSPC)的自体移植可以在不久的将来作为选择的多种遗传疾病(包括溶酶体储存疾病(LSD))的选择。HSPC的传统基因治疗方法基于慢病毒载体对转基因的整合,而最近靶向的盒式磁带的整合通常由设计器核酸酶支持。无论哪种情况,转基因的表达通常都由外源无处不在的启动子维持,该启动子可能会改变或失调周围的原始癌基因和/或肿瘤抑制器的表达。此外,普遍存在的启动子在干细胞水平上诱导所需转基因的表达,这可能会影响其功能性,因为已建议将半乳糖脑脑苷酶(Krabbe)或葡萄糖脑苷酶(Gaucher)的过表达表达。,我们基于将剪接功能的盒式集成到谱系特异性基因座的内含子中的集成基于HSPCS的新型基因编辑系统。这种方法旨在防止转基因在干细胞水平上的表达,仅在细胞分化后触发转基因表达。作为概念证明,我们编辑了CD11b在HSPC中的内含子,并诱导转基因(用于治疗I型I型的GFP或IDUA)在体外分化和体内髓细胞塑料插入后的髓样含量I型)中的髓样特异性表达。我们证明了这种方法对CD20和CD4基因的可运输能力。

碳水化合物代谢遗传疾病中的代谢性心肌病和心脏缺陷:系统评价。 Conte,F。; Sam,J.E。; Lefeber,
摘要:心力衰竭(HF)是一种进行性慢性病,仍然是全球死亡的主要原因,影响了6400万以上的患者。HF可能是由具有单基因病因的心肌病和先天性心脏缺陷引起的。与心脏缺陷发展相关的基因和单基因疾病的数量正在不断增长,并包括遗传的代谢杂志(IMD)。已经报道了几种影响各种代谢途径的IMD,出于心肌病和心脏缺陷。考虑到糖代谢在心脏组织中的关键作用,包括能量产生,核酸合成和糖基化,与心脏表现相关的越来越多的与碳水化合物代谢相关的IMD越来越多。在这项系统的综述中,我们提供了与碳水化合物代谢相关的IMD的全面概述,这些IMD呈现出心肌病,心律失常疾病和/或结构性心脏缺陷。我们识别出患有心脏并发症的58 IMD:3糖/糖连接转运蛋白的缺陷(GLUT3,GLUT10,THTR1); 2个磷酸盐途径的疾病(G6PDH,TALDO); 9糖原代谢疾病(GAA,GBE1,GDE,GYG1,GYS1,LAMP2,RBCK1,PRKAG2,G6PT1); 29 congenital disorders of glycosylation (ALG3, ALG6, ALG9, ALG12, ATP6V1A, ATP6V1E1, B3GALTL, B3GAT3, COG1, COG7, DOLK, DPM3, FKRP, FKTN, GMPPB, MPDU1, NPL, PGM1, PIGA, PIGL, PIGN, PIGO,PIGT,PIGV,PMM2,POMT1,POMT2,SRD5A3,XYLT2); 15碳水化合物连接的溶酶体储存疾病(CTSA,GBA1,GLA,GLB1,HEXB,IDUA,IDS,IDS,SGSH,NAGLU,HGSNAT,GNS,GNS,GALNS,GALNS,GALNS,ARSB,ARSB,GUSB,GUSB,ARSK)。通过这项系统评价,我们旨在提高人们对碳水化合物连接IMD的心脏介绍的认识,并引起人们对碳水化合物连接的致病机制的注意,这些致病机制可能是心脏并发症的基础。