XiaoMi-AI文件搜索系统
World File Search SystemIFNS
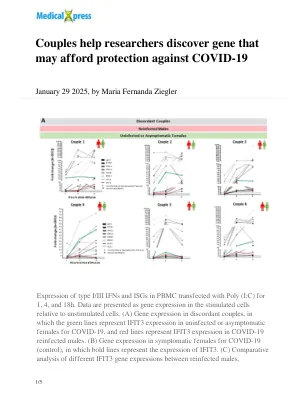
夫妇帮助研究人员发现可能能够保护Covid-19
I/III型IFN和ISG在PBMC中用Poly(I:C)转染为1、4和18H的表达。数据相对于未刺激的细胞,将数据作为基因表达表示。(a)不一致的夫妻中的基因表达,其中绿线代表未感染或无症状的女性在covid-19中表示IFIT3的表达,而红线表示在Covid-19的COVID-19再感染的雄性中的IFIT3表达。(b)在症状女性中的基因表达于19(对照),其中粗线表示IFIT3的表达。(c)对重新感染的雄性之间不同IFIT3基因表达的比较分析,

RNF138通过抑制IRF3激活来下调抗病毒先天免疫 对Monkeypox(MPOX)病毒抗原免疫的血清监管
摘要:一种病毒感染激活转录因子IRF3和NF-κB,它们协同诱导I型干扰素(IFNS)。在这里,我们将E3泛素连接酶RNF138鉴定为病毒触发的IRF3激活和IFN-β诱导的重要负调节剂。RNF138的过表达抑制了病毒诱导的IRF3激活和IFNB1基因的转录,而RNF138的敲除促进了病毒诱导的IRF3的激活和IFNB1基因的转录。我们进一步发现,RNF138促进了PTEN的泛素化,随后抑制了PTEN与IRF3的相互作用,这对于PTEN介导的IRF3的核易位至关重要,从而抑制IRF3进口到核中。我们的发现表明,RNF138通过抑制PTEN与IRF3的相互作用来负调节病毒触发的信号传导,这些数据为细胞抗病毒反应的分子机制提供了新的见解。

13 种 IFN-α 亚型的异质性和功能
I 型 IFN 对宿主对病毒感染的反应至关重要,也与多种自身免疫性疾病的发病机制有关。I 型 IFN 家族中有多种亚型,特别是 13 种不同的 IFN-α 基因,它们通过哺乳动物细胞中普遍表达的相同异二聚体受体发出信号。进化遗传学研究和功能性抗病毒检测都强烈表明 13 种 IFN-α 亚型之间存在不同的功能和活性,但我们仍然对这些不同的作用缺乏清晰的认识。本综述总结了描述 IFN-α 亚型不同功能的研究证据,并强调了报告之间存在差异的潜在原因。我们研究了急性和慢性病毒感染以及自身免疫,并整合了最近对抗 IFN-α 自身抗体在这些不同情况下塑造 I 型 IFN 反应的重要性的认识。

乳杆菌Delbrueckii Letm
L. delbrueckii le tm的delbrueckii形式具有出色的免疫调节特性。它通过非特异性和特定的联系影响先天和适应性免疫,从而根据受试者的免疫状态来控制Th1和Th2途径对免疫反应的协调。通过刺激关键细胞因子IFN,TNF,NK细胞,IL-1,IL-2和IL-6的产生来诱导特定的抗体产生并平衡人免疫系统的能力,以保护感染和癌细胞。它也是一种免疫调节剂,可以通过细胞介导的免疫来平衡和归一化的非特异性反应,并在病原剂存在下诱导更高的反应。它在慢性疾病,炎症和免疫失速(如肿瘤)中提供了免疫平衡(通过抑制)。LE菌株因此调节了对内源性和外源致病剂的免疫反应。

MDA5依赖性响应 - to-tribute-to-AutoMune- ...
引言1型糖尿病(T1D)是T细胞介导的自身免疫性疾病,导致胰腺β细胞破坏(1)。提出了遗传学,环境和免疫系统的协同作用来诱导T1d(2-5)。单子双胞胎的T1D具有约30%–50%的一致性,这表明环境在T1D发育中起着重要作用(6,7)。与T1D相关的一个环境因素是Coxsackievivirus B(CVB)感染(8,9)。CVB病毒RNA和/或病毒颗粒已在最近发作T1D的患者的血液,粪便和胰岛中检测到(9-11)。在非肥胖糖尿病(NOD)小鼠模型中,CVB感染通过诱导炎症性胰腺抗病毒反应加速T1D,导致β细胞破坏(12,13)。由IFIH1基因编码的先天病毒传感器分化相关蛋白5(MDA5)检测到DSRNA病毒复制中间体并启动抗病毒信号传导(14,15)。MDA5结合其配体后的关键反应之一是I型IFN的合成,例如IFN-α和IFN-β,以促进巨噬细胞,树突状细胞和T细胞的病毒清除和激活(16-20)。尽管I型IFN对抗病毒反应至关重要,但它们也与早期T1D发育有关(21,22)。在转基因CD1小鼠中,其中β细胞组成型表达IFN-α,T1D发作发生在10周龄的60%小鼠中(23)。相反,在NOD雌性小鼠中,IFN -α和-β受体亚基1(IFNAR1)表达的丧失导致T1D发育显着延迟(24)。在T1D患者中,在自身抗体发育之前在血液中检测到I型IFN基因特征(21、22),而GWAS发现与I型IFN合成和信号传导有关的T1D基因,例如IFIH1(例如IFIH1)(25,26)。IFIH1中的多个单核苷酸多态性(SNP)与人类T1D发育有关。A946T SNP(rs1990760)在氨基酸946时导致丙氨酸对硫代的变化,与T1D风险相关,并导致IFN-α /β和IFN刺激的基因产生< / div>的增加

释放系统性 STING 激动剂在癌症免疫治疗中的潜力
先天免疫是启动和维持适应性免疫反应的关键[1]。抗肿瘤免疫反应也不例外,它也依赖于先天免疫系统来提供强大而持久的免疫反应。过去十年,越来越多的证据表明,环状 GAM-AMP 合酶 (cGAS)-干扰素基因刺激物 (STING) 通路是癌症免疫中一条关键的先天免疫激活通路 [2,3]。简而言之,先天免疫细胞通过细胞内的 cGAS 检测肿瘤衍生的 DNA,从而催化环状 GAM-AMP (cGAMP) 的生成。细胞浆 cGAMP 激活 STING 并诱导 I 型 IFN 以及其他促炎细胞因子,从而协调抗肿瘤免疫。药理学激活 STING 已被证明是各种临床前模型中有效的癌症免疫疗法[4]。第一代 STING 激动剂已在临床试验中进行评估,包括 ADU-S100 和 MK-1454。两者都是基于环二核苷酸 (CDN) 的化合物,可直接注射到肿瘤中。

第56届中西部学生生物医学研究论坛
背景,意义和假设:沙眼衣原体(CTR)是一种强制性细胞内病原体,是细菌性传播感染(STI)的主要原因。尽管通常无症状,但感染可能会发展为上等生殖道,并可能导致严重的生殖健康后遗症,例如骨盆炎性疾病,异位妊娠,甚至不育(如果未经治疗)。全国有160万例案件,直接终身费用超过6.9亿美元,CTR被认为是主要的公共卫生负担。一些人自发清除感染,这些感染归因于宿主的适应性免疫。然而,研究还表明,感染可能会持续存在,并重新感染表明长期保护性免疫充其量是部分部分。尽管对CTR感染的适应性免疫反应已充分表征,但主动感染如何影响宿主的先天免疫力,尤其是在CTR-上皮界面上仍未开发。此外,通常会忽略存在未感染的旁观者细胞的宫颈上皮感染期间宿主反应的表征,而不是使用完全感染的上皮单层感染模型,其中在单个时间点感染后在单个时间点收集样品。这进一步强调了在感染过程中调查跨多个时间点的宿主反应的需求,这可能对CTR存活和扩散有影响。宫颈上皮位于CTR -HOST相互作用的中心,因为它是感染的主要部位。可溶性因子在内,包括干扰素(IFN)在感染微环境中产生丰富的。上皮相关的IFN(例如IFNβ和IFNλ)以自分泌和旁分泌方式通过JAK-STAT途径驱动IFN刺激的基因(ISGS)的表达。由于大多数ISG是推定的抗菌剂,因此累积上皮反应通常是抗菌剂,有助于病原体限制。因此,感染上皮细胞中的细胞因子信号传导通常是颠覆病原体的目标。我们最近表明,CTR可以抑制完全感染的上皮单层的上皮IFN反应。这与该领域所谓的CTR上皮宿主免疫生物学相反,通常被视为促炎性。为了调和观察到的减毒上皮IFN与当前炎症性CTR上皮相互作用的概念,我们假设旁观者细胞对于塑造细胞因子环境至关重要,并且在塑造上皮IFN反应的失调可能对CTRENSINCAINS产生导致的影响。

激活内源性逆转录病毒和胰腺癌中MEK1/2抑制病毒模仿的诱导
虽然胰管导管腺癌(PDACS)沉迷于KRAS激活突变,但下流kras效应子的抑制剂,例如MEK1/2激酶抑制剂TRAMETINIB,却没有治疗作用。但是,由KRAS途径衰减驱动的监管电路的广泛重新布线可能会引起治疗相关性的脆弱性。在MEK1/2通过Trametinib抑制后的最初几个小时,对PDAC细胞中的转录和表观基因组变量进行了深入的分子分析,揭示了诱导内组逆转录病毒(ERV)(ERVS)的诱导,从而逃脱了表观遗传的硅烷,从而产生了双链RNAS和Interfecn of interfece and Interfecron的生产(导致了Interfef)(Interfe)的产生。我们跟踪了ERV激活,以早期诱导量写因子ELF3的早期诱导,该因子ELF3在IFN和IFN刺激的基因的激活中与IRF1(干扰素调节因子1)进行了广泛结合和激活。在免疫肿瘤学中合理设计中,可以利用 trametinib诱导的PDAC中的病毒模仿。

rop16通过通过刺激的多泛素化
摘要:CGAS刺信信号传导是诱导I型IFN的主要途径,在防御巨型T. gondii感染中起着至关重要的作用。相比之下,T。Gondii制定了多种策略来抵消宿主防御,从而在广泛的宿主中引起严重疾病。在这里,我们证明了T. gondii Rhoptry蛋白16(ROP16)通过抑制CGA(环状GMP-AMP合酶)途径通过刺痛的多素化抑制I型干扰素信号传导。Mech-在动态上,ROP16通过信号域与STING相互作用,并抑制NLS(核定位信号)domain依赖性方式中STIN的K63连接的泛素化。conse,在Pru tachyzoites中淘汰了ROP16,促进了I型IFN的刺激介导的产生,并限制了T. gondii的复制。一起,这些发现描述了一种独特的途径,其中T. gondii利用了sting的泛素化来逃避宿主的抗寄生虫免疫,从而揭示了对宿主与寄生虫之间相互作用的新见解。

单细胞水平的病毒感染与先天免疫之间的对抗
被病毒感染时,细胞可能会分泌干扰素(IFN),该干扰素(IFN)促使附近细胞为即将到来的感染做准备。相互,病毒蛋白通常会干扰IFN合成和IFN诱导的信号传导。我们使用基于药物的随机方法对传播病毒与先天免疫反应之间的串扰进行了建模。通过分析免疫荧光显微镜图像,我们观察到呼吸综合病毒(RSV)(RSV)和感染A549细胞之间的相互拮抗作用会导致单细胞水平和复杂的细胞信号传导状态空间模式的二分法反应。我们的分析表明RSV在三个层面上阻止了先天反应:通过抑制IRF3激活,抑制IFN合成以及抑制STAT1/2激活。反过来,由IFN刺激(STAT1/2激活)基因编码的蛋白质抑制了病毒RNA和病毒蛋白的合成。这些抑制作用的显着结果是病毒蛋白缺乏巧合和单个细胞中IFN的表达。该模型可以研究免疫刺激有缺陷的病毒颗粒和信号网络扰动的影响,这些影响可能有可能促进病毒感染的遏制或清除。