XiaoMi-AI文件搜索系统
World File Search SystemKo

中链脂肪酸通过免疫调节受体GPR84
1型糖尿病(T1D)是一种自身免疫性疾病,导致胰腺β细胞破坏。coxsackievivirus B3(CVB3)感染和黑色素瘤分化相关蛋白5依赖性(依赖MDA5)抗病毒反应与T1D发育有关。IFIH1中的突变(编码为MDA5)与T1D易感性相关,但是这些突变如何促进T1D尚不清楚。Utilizing nonobese diabetic (NOD) mice lacking Ifih1 expression ( KO ) or containing an in-frame deletion within the ATPase site of the helicase 1 domain of MDA5 (Δ Hel1 ), we tested the hypothesis that partial or complete loss-of-function mutations in MDA5 would delay T1D by impairing proinflammatory pancreatic macrophage and T cell responses.在雌性点头和KO小鼠中开发的自发T1D类似,但在δHEL1小鼠中显着延迟,这可能部分是由于髓样衍生的抑制细胞同时增加。有趣的是,与点头小鼠相比,KO雄性小鼠自发性T1D增加了。虽然点头和KO小鼠产生了CVB3加速的T1D,而δHEL1小鼠则部分是由于I型IFN,胰腺浸润TNF +巨噬细胞,IFN-γ + CD4 + T细胞和perforin + CD8 + T细胞的部分保护。 此外,与野生型MDA5相比,δHEL1 MDA5蛋白减少了ATP水解。 我们的结果表明,MDA5功能受阻会延迟T1D,但MDA5的损失促进了T1D。虽然点头和KO小鼠产生了CVB3加速的T1D,而δHEL1小鼠则部分是由于I型IFN,胰腺浸润TNF +巨噬细胞,IFN-γ + CD4 + T细胞和perforin + CD8 + T细胞的部分保护。此外,与野生型MDA5相比,δHEL1 MDA5蛋白减少了ATP水解。我们的结果表明,MDA5功能受阻会延迟T1D,但MDA5的损失促进了T1D。

MDA5依赖性反应有助于自身免疫性糖尿病进展和阻碍
1型糖尿病(T1D)是一种自身免疫性疾病,导致胰腺β细胞破坏。coxsackievivirus B3(CVB3)感染和黑色素瘤分化相关蛋白5依赖性(依赖MDA5)抗病毒反应与T1D发育有关。IFIH1中的突变(编码为MDA5)与T1D易感性相关,但是这些突变如何促进T1D尚不清楚。Utilizing nonobese diabetic (NOD) mice lacking Ifih1 expression ( KO ) or containing an in-frame deletion within the ATPase site of the helicase 1 domain of MDA5 (Δ Hel1 ), we tested the hypothesis that partial or complete loss-of-function mutations in MDA5 would delay T1D by impairing proinflammatory pancreatic macrophage and T cell responses.在雌性点头和KO小鼠中开发的自发T1D类似,但在δHEL1小鼠中显着延迟,这可能部分是由于髓样衍生的抑制细胞同时增加。有趣的是,与点头小鼠相比,KO雄性小鼠自发性T1D增加了。虽然点头和KO小鼠产生了CVB3加速的T1D,而δHEL1小鼠则部分是由于I型IFN,胰腺浸润TNF +巨噬细胞,IFN-γ + CD4 + T细胞和perforin + CD8 + T细胞的部分保护。 此外,与野生型MDA5相比,δHEL1 MDA5蛋白减少了ATP水解。 我们的结果表明,MDA5功能受阻会延迟T1D,但MDA5的损失促进了T1D。虽然点头和KO小鼠产生了CVB3加速的T1D,而δHEL1小鼠则部分是由于I型IFN,胰腺浸润TNF +巨噬细胞,IFN-γ + CD4 + T细胞和perforin + CD8 + T细胞的部分保护。此外,与野生型MDA5相比,δHEL1 MDA5蛋白减少了ATP水解。我们的结果表明,MDA5功能受阻会延迟T1D,但MDA5的损失促进了T1D。

CRISPR/Cas9 的转录组学和蛋白质组学分析...
摘要:Arc/Arg3.1(活性调节细胞骨架相关蛋白(ARC))是长期突触可塑性的关键调节器,并参与精神分裂症的病理生理。人类 ARC 作用的功能和机制尚不清楚,值得进一步研究。为了在体外研究 ARC 基因的功能,我们通过 CRISPR/Cas9 介导的基因编辑生成了 ARC 敲除 (KO) HEK293 细胞系,并进行了 RNA 测序和非标记 LC-MS/MS 分析,以识别同源 ARC -KO HEK293 细胞中差异表达的基因和蛋白质。此外,我们使用生物发光共振能量转移 (BRET) 分析来检测 ARC 蛋白与差异表达蛋白之间的相互作用。ARC 的基因缺失会扰乱参与细胞外基质和突触膜的多个基因。发现 ARC -KO 细胞和 ARC 野生型细胞之间存在 7 种蛋白质(HSPA1A、ENO1、VCP、HMGCS1、ALDH1B1、FSCN1 和 HINT2)的差异表达。BRET 测定结果表明 ARC 与 PSD95 和 HSPA1A 相互作用。总体而言,我们发现 ARC 调节涉及细胞外基质、突触膜和热休克蛋白家族的基因的差异表达。本文介绍的 ARC -KO HEK293 细胞的转录组和蛋白质组学谱为 ARC 作用的潜在机制和涉及精神分裂症病理生理的分子通路提供了新的证据。

slitrk4是在外侧杏仁核的恐惧记忆回路中发展抑制性神经元所必需的
Slitrk家族由六个突触粘附分子组成,其中一些分子与神经精神疾病有关。在这项研究中,我们旨在通过分析slitrk4敲除(KO)小鼠来研究slitrk4的生理作用。SLITRK4蛋白在大脑中被广泛检测到,并且在嗅球和杏仁核中很丰富。在系统的行为分析中,男性slitrk4 ko小鼠在对经典恐惧条件的提示测试中表现出增强的恐惧记忆,而社会行为在相互的社交互动测试中表现出来。在使用杏仁核切片的电生理分析中,slitrk4 ko小鼠在丘脑 - 杏仁核的长期增强率增强,并减少了反馈抑制。在SLITRK4 KO大脑的分子标记分析中,成人阶段的侧杏仁核前部减少了钙网蛋白(CR)阳性中间神经元的数量。在体外实验中,在神经元之间的实验中,Slitrk4降低的胚胎干细胞在诱导GABA能中间神经元中有缺陷,其对Sonic HedgeHog信号激活的响应改变了GABA> GABA> GABA> GABA> GABAERNERNERORON子集。这些结果表明SLITRK4功能与恐惧记忆回路中抑制性神经元的发展有关,并将有助于更好地理解骨质应激障碍,在这种障碍中,已经报道了SLITRK4的表达改变。

近端小管缩短对Lowe综合征模型中蛋白质排泄的影响
底物是内吞作用的主要调节剂,预计LS LS患者的LMW蛋白尿是由于PT顶端内吞途径沿PT的某些有效功能所致。3与此相一致,培养细胞模型中的一部分研究表明,OCRL在内吞回收中起作用,这是通过防止在内吞囊泡上积累的肌动蛋白涂层的解聚和/或回收箱的作用。4,5但是,OCRL在细胞稳态中也具有许多其他角色,包括睫状生物发生,6-8细胞极性和自噬。6,9,10此外,OCRL在细胞因子期间被招募到脱落部位。11 ptdins(4,5)p 2累积稳定在细胞因子过程中的细胞内桥,并且其通过OCRL的水解对于脱落是必要的。11尚不清楚这些功能如何促进LS病理学。另一个未解决的问题是,OCRL的损失如何损害LS患者的Ca 2+,HCO 3 2和氨基酸的PT恢复。近年来已经开发了LS的小鼠和斑马鱼模型,但是在细胞培养中观察到的分子和细胞缺陷与患者和动物模型的表型之间的联系仍然难以捉摸。缺乏OCRL的转基因斑马鱼表现出降低的巨蛋白水平,降低了流体相位标记物的上升水平,除了与LS患者观察到的患者一致的眼睛和面部缺陷外,促脑肾脏PT中的亚皮囊泡较少。8,12小鼠LS模型的开发更为复杂。 这些8,12小鼠LS模型的开发更为复杂。这些OCRL敲除(KO)小鼠没有明显的表型,因为它们表达了高水平的Inpp5b,这是另一种磷脂酰肌醇5 9-磷酸酶,显然可以对某些OCRL功能进行操作。13 - 15小鼠PT中的inpp5b在较高水平和与人类相比的剪接变体中表达不同。16由于小鼠中的OCRL和INPP5B的全局KO是致命的,因此通过跨越OCRL KO小鼠的OCRL KO小鼠产生了17,18 LS小鼠模型,该小鼠过表达人Inpp5b与小鼠INPP5B KO:由此产生的雄性小鼠在年龄的8周时表现出适中的蛋白尿和氨基尿症。19,20已描述了一个最近的小鼠模型,其中在OCRL KO小鼠的肾脏中有条件地灭活了INPP5B。这些小鼠中的21个PT细胞表达了巨蛋白水平降低,并且表现出严重受损的内吞作用。令人惊讶的是,在KO之后没有立即观察到蛋白尿,而是需要几个月的发展。此时间滞后与OCRL对内吞途径功能的直接影响不一致,并表明在更长的时间段内发生的其他变化与LS表型相关。此外,需要靶向OCRL和INPP5B以观察任何肾脏表型,这是努力确定OCRL在Pt功能中的特定作用的努力。为了研究OCRL的损失如何影响PT功能,我们产生了PT细胞中LS的慢性CRISPR/CAS9 OCRL KO和LS的急性siRNA敲低模型。引人注目的是,在我们的所有模型中以及在患者纤维细胞中,我们观察到功能性OCRL的损失延长了细胞分裂的持续时间,并导致了多核细胞的积累。


视频:辅助喂养机器人在实验室外进行测试
在两项研究中,用户都成功地养活了自己的饭菜。在第一项研究中,机器人获得了约80%精度的主菜,另一项研究中的用户发现这是成功的阈值。在第二次研究中,房屋的各种环境和环境(Ko)可能在低光或在床上工作时在饮食中吃饭 - 使系统的默认功能保持不变。但研究人员将该系统设计为可定制的,因此KO能够控制机器人并仍然为自己喂食所有餐点。
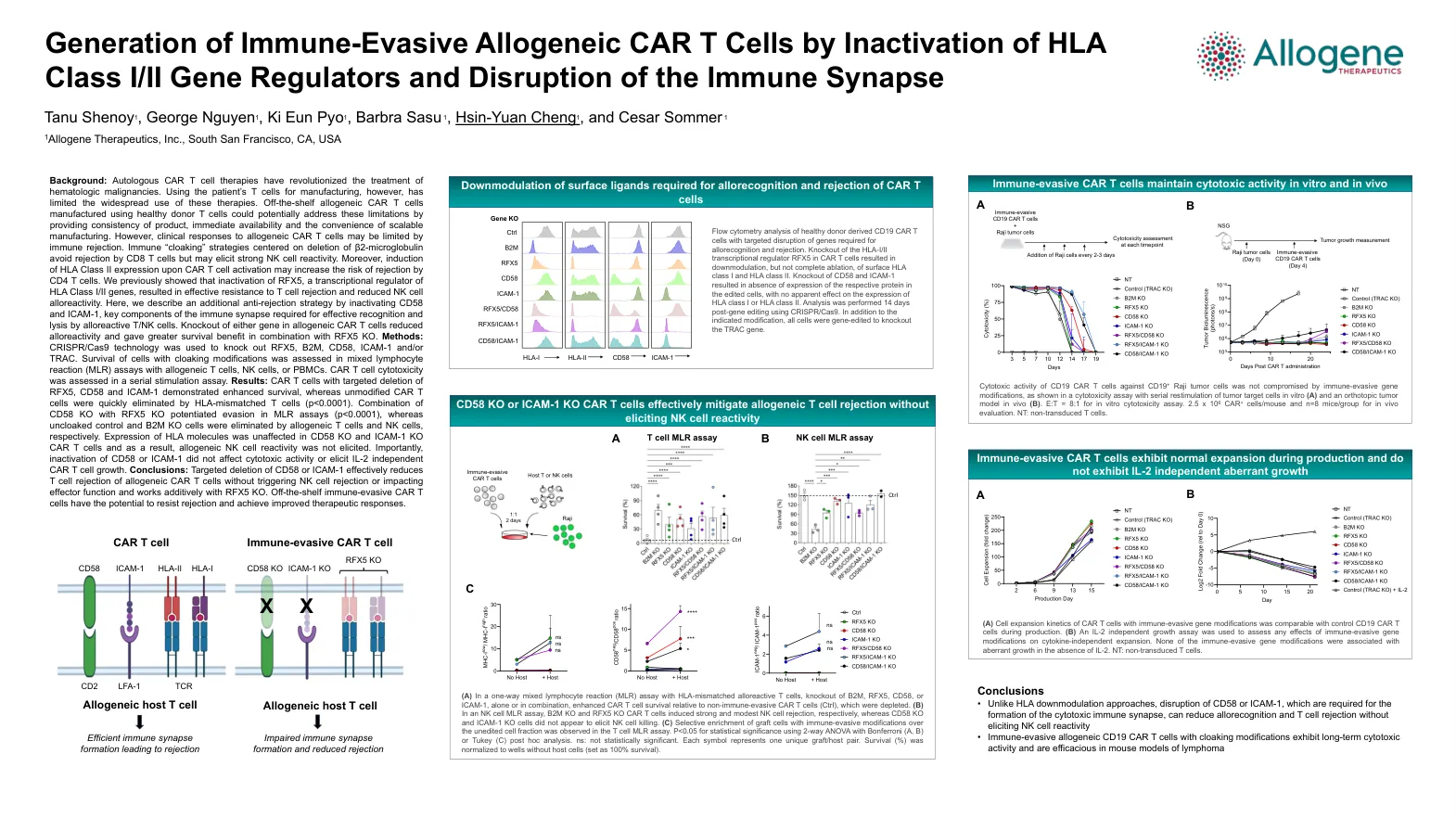
通过失活 HLA I/II 类基因调节剂和破坏免疫突触来生成免疫逃避同种异体 CAR T 细胞
背景:自体 CAR T 细胞疗法彻底改变了血液系统恶性肿瘤的治疗。然而,使用患者的 T 细胞进行制造限制了这些疗法的广泛使用。使用健康供体 T 细胞制造的现成同种异体 CAR T 细胞可能通过提供产品的一致性、即时可用性和可扩展制造的便利性来解决这些限制。然而,对同种异体 CAR T 细胞的临床反应可能受到免疫排斥的限制。以删除 β2-微球蛋白为中心的免疫“隐身”策略避免了 CD8 T 细胞的排斥,但可能引起强烈的 NK 细胞反应性。此外,CAR T 细胞活化后诱导 HLA II 类表达可能会增加 CD4 T 细胞排斥的风险。我们之前表明,RFX5(HLA I/II 类基因的转录调节因子)的失活可有效抵抗 T 细胞排斥并降低 NK 细胞同种异体反应性。在这里,我们描述了一种额外的抗排斥策略,即失活 CD58 和 ICAM-1,它们是同种反应性 T/NK 细胞有效识别和溶解所需的免疫突触的关键组成部分。同种异体 CAR T 细胞中任一基因的敲除均降低了同种异体反应性,并与 RFX5 KO 结合使用可带来更大的生存益处。方法:使用 CRISPR/Cas9 技术敲除 RFX5、B2M、CD58、ICAM-1 和/或 TRAC。在使用同种异体 T 细胞、NK 细胞或 PBMC 的混合淋巴细胞反应 (MLR) 测定中评估了具有隐形修饰的细胞的生存率。在连续刺激测定中评估了 CAR T 细胞的细胞毒性。结果:靶向删除 RFX5、CD58 和 ICAM-1 的 CAR T 细胞表现出增强的生存率,而未经修饰的 CAR T 细胞很快被 HLA 不匹配的 T 细胞消灭(p<0.0001)。 CD58 KO 与 RFX5 KO 的组合增强了 MLR 测定中的逃避(p<0.0001),而未隐藏的对照和 B2M KO 细胞分别被同种异体 T 细胞和 NK 细胞消除。CD58 KO 和 ICAM-1 KO CAR T 细胞中的 HLA 分子表达不受影响,因此不会引发同种异体 NK 细胞反应性。重要的是,CD58 或 ICAM-1 的失活不会影响细胞毒活性或引发 IL-2 独立的 CAR T 细胞生长。结论:靶向删除 CD58 或 ICAM-1 可有效降低同种异体 CAR T 细胞的 T 细胞排斥,而不会触发 NK 细胞排斥或影响效应功能,并与 RFX5 KO 协同作用。现成的免疫逃避 CAR T 细胞具有抵抗排斥和实现改善治疗反应的潜力。

癌症中与心血管健康相关的生活质量
SLC16A2-G401R和基因敲除(KO)细胞系是通过转染重组Cas9蛋白和(G401 MUT)的合成GRNA或没有SSODN的HIPSC系列而产生的,从健康的供体(Bibhi001-b)中产生的HIPSC系。使用单个GRNA靶向外显子3生成了两种修饰(图1 A,表2)。SLC16A2-G401R Mu tation是通过使用SSODN模板通过同源性修复(HDR)引入的(图1 A,表2)。SSODN模板包括在AHDS患者中发现的突变:C.1201G> A,静音突变,以破坏PAM序列:C.1200C> T和4个无声突变,以破坏种子序列(C.1185C> T,C.1186C> A,C.1188C> A,C.1188C> A,C.1191C> a,C.1191c>1 a和f)。使用了相同的GRNA指南,并选择了带有框架移动的克隆,从而选择了过早的停止密码子。在两个报道的KO细胞系中(BIHI001-B-7和 - 8)

参考文献Agrahar-Murugkar,D.,Gulati,P.,Kotwaliwale ...
bud ad aki,S.,Kocevakomleni®,D.,Lukinacčić,J. KožUu ul,Ž。 div>(2014)。 div>