XiaoMi-AI文件搜索系统
World File Search SystemLDT
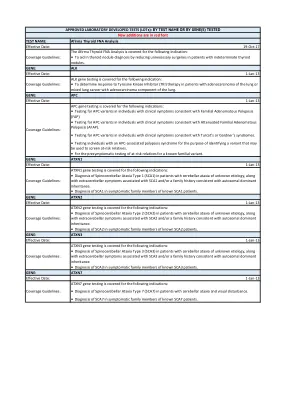
TRICARE 批准的 LDT 列表.xlsx
BCR/ABL1 基因检测涵盖以下适应症:• 通过定量 RT PCR (RQ-PCR) 对疑似慢性粒细胞白血病 (CML) 患者进行诊断评估。• 通过定性 RT-PCR 对疑似 CML 患者进行诊断评估。• 通过 RQ-PCR 监测 CML 患者对 TKI 疗法(如伊马替尼)的反应。• 检测 CML 患者中是否存在 BCR/ABL1 p.Thr315Ile 变异,以指导对一线伊马替尼疗法产生耐药性后的治疗选择。• 检测 CML 患者中是否存在除 p.Thr315Ile 以外的 BCR/ABL1 变异,以指导对一线伊马替尼疗法产生耐药性后的治疗选择。检测名称:生物治疗学乳腺癌指数生效日期:2023 年 1 月 1 日生物治疗学乳腺癌指数涵盖以下适应症:

CD-2021-12 – 修订版 (LDV, LDT) 主题
• 车辆说明书应明确说明如何将车辆置于测功机模式(见下文)、如何将车辆置于空档、所需的电流钳数量以及如何安装它们以及如何读取电压。制造商必须包括测试车辆独有的任何特殊说明。这可能包括如何使用历史上称为“钥匙”的东西、如何“启动”车辆以及如何使车辆进入“休眠”状态。任何可能干扰测试的项目,如车门打开、引擎盖打开或安全带解开时车辆自动禁用,都应详细说明和强调,这些项目无法通过测功机模式或其他方式关闭。 • 车辆说明书应包括在 MCT 的两个恒速部分上实现的预期时间和距离。如果没有提供这些值,EPA 可能会使用 SAE J1634-12 中的公式为测试中期恒速循环 (CSC M ) 和测试结束时恒速循环 (CSC E ) 设置预期距离。 • 车辆必须有 CD-16-03 中所述的用于底盘测功机测试的固定装置。 • EPA 必须使用 EPA 自己的电流钳进行电流测量。 • 车辆应有清晰标记的电流钳连接位置和电压抽头(如果使用电压抽头)。车辆说明应包括安全安装车辆电流钳的详细说明。制造商必须指明电流流动的方向以及他们希望如何安装电流钳。EPA 实验室现在使用电流惯例,即负电流流出电池(放电)和正电流流入电池(充电)。 • EPA 更喜欢在电压抽头上使用 Pomona #6383-02 连接(通常称为“带护套的香蕉插头”)。如果由于技术原因您无法安装这种类型的连接,请通知您的认证工程师。 • 不提供电压抽头并需要 CAN 数据采集来测量电压的车辆可能会导致测试数据计算延迟。制造商应在预期结果时间中额外预留最多一周的时间。如果制造商可以提供包含参数信息的 .dbc 文件,EPA 能够使用其测试单元控制器读取 CAN 消息。如果制造商将提供第三方数据记录器,则他们必须提供电池电压、车速和时间对齐数据,然后才能提交给实验室进行处理。• 如果您的车辆需要四个以上的电流钳,请在测试前告知您的认证工程师。在这些情况下,制造商还应提供有关如何在测试数据采集中使用不同电流钳的详细说明,例如加法、减法等。• 如果需要起重机来安装电流钳或电压接头,请在测试前告知您的认证工程师。请告知是否需要任何特殊工具


ld-肽对农杆菌的适应性和极性生长至关重要
肽聚糖(PG)是一种网状结构,是细菌细胞壁的主要成分,对于维持细胞完整性和形状至关重要。大多数细菌依靠青霉素结合蛋白(PBP)进行交联,但某些物种也采用LD-转肽酶(LDTS)。与PBP不同,LDT的本质和生物学功能在很大程度上不清楚。以其极性生长而闻名的字母细菌的杂种菌序,其PG异常富含LD-Crosslinks,这表明LDT在这些细菌中可能在PG合成中起更重要的作用。在这里,我们研究了植物病原体农杆菌tumefaciens中的LDT,发现该细菌中至少有14个假定的LDT中的14种引起的LD-肽对其存活至关重要。值得注意的是,缺乏独特的7个LDT的突变体在杂种菌中广泛保守的突变体表现出降低的LD互动和PG将PG束缚到外膜β-贝尔β-桶蛋白上的链接。因此,这种突变体遭受了严重的健身损失和细胞形状的圆形,强调了这些菌粒特异性LDT在维持细胞壁完整性和促进延伸方面所起的关键作用。tn-sequering屏幕表现出了a的非冗余功能。Tumefaciens LDTS。具体而言,连字符特异性LDTs与除法和细胞周期蛋白表现出合成的遗传相互作用,而来自另一组的单个LDT。此外,我们的发现表明,缺乏所有LDT的菌株表现出独特的表型特征和遗传相互作用。总体而言,我们的工作强调了ld-rosslinking在a中的关键作用。tume-faciens细胞壁完整性和生长,并为这些交联活动的功能专业化提供了见解。

慢性淋巴细胞白血病:2022 年诊断和治疗程序更新
4. 进行性淋巴细胞增多,2 个月内增多≥50%,或淋巴细胞倍增时间(LDT)小于 6 个月。LDT 可通过在 2-3 个月的观察期内每隔 2 周获得的绝对淋巴细胞计数(ALC)的线性回归外推获得;初始血液淋巴细胞计数<30 000/μL 的患者可能需要更长的观察期来确定 LDT。应排除除慢性淋巴细胞白血病之外的其他导致淋巴细胞增多的因素(例如感染、类固醇给药)



实验室开发的测试:小实体合规指南
2024年5月6日,FDA在题为“医疗设备;实验室开发了测试”的联邦公报上发布了最终规则(89 FR 37286)(“ LDT最终规则”)。该最终规则修改了FDA法规,以明确表明体外诊断产品(IVD)是《联邦食品,药物和化妆品法》(FD&C ACT),包括IVD制造商是实验室的设备。1该修正案反映出FD&C ACT中的设备定义并未区分制造设备的实体。与修正案结合使用,FDA逐步淘汰其对实验室开发的测试(LDTS)2的一般执行酌处权方法,以便实验室制造的IVD通常与其他IVD相同的执法方法(即FDA对合规性的期望通常相同)。在LDT最终规则的序言中,更全面地描述了该阶段的策略,其中包括针对由实验室制造的特定类别的IVD的执法酌处权,包括目前以LDTS 3和LDTS提供的IVD,以满足未满足的需求。

新墨西哥州监测和跟踪测试...
• 该检测尚未获得 FDA 批准或认可; • 该检测已获得 FDA 的 EUA 授权,可供授权实验室使用; • 该检测仅被授权用于检测 SARSCoV-2 核酸,不适用于任何其他病毒或病原体;并且, • 该检测仅在声明存在情况证明根据该法案第 564(b)(1) 节、21 USC § 360bbb-3(b)(1) 授权紧急使用体外诊断试剂检测和/或诊断 COVID-19 期间被授权,除非授权提前终止或撤销。 6. 什么是灵敏度和特异性? 在医学诊断中,检测灵敏度是检测正确识别患有疾病的人的能力(真阳性率),而检测特异性是检测正确识别未患有疾病的人的能力(真阴性率)。 7. Quest LDT、Hologic 和 Roche 检测的灵敏度/特异性是多少?对于分子检测,我们使用 Quest LDT、Roche 和 Hologic 检测。FDA EUA 网站包含所有 4 种分子检测的制造信息。检测的敏感性和特异性列在每条记录下链接的使用说明 (IFU) 文件中。制造商有责任为其检测提供此信息。以下是 IFU 文件的链接。

CB&CDx2024
新鲜与创新:了解 ADC 中的 CDx、LDT 的最新动态、生成式 AI 和无监督学习模型。此外,我们将探索变革性液体生物标记物、非侵入性数字生物标记物和非肿瘤生物标记物。这些见解将使您能够在产品线中实施重大变革,从而为所有患者提供精准医疗。