XiaoMi-AI文件搜索系统
World File Search SystemLigand

CISH 的基因编辑可独立于免疫检查点细胞表面配体的表达增强 T 细胞效应程序
。CC-BY-NC-ND 4.0 国际许可,根据 (未经同行评审认证)提供,是作者/资助者,他已授予 bioRxiv 永久展示预印本的许可。它是此预印本的版权持有者,此版本于 2021 年 8 月 17 日发布。;https://doi.org/10.1101/2021.08.17.456714 doi:bioRxiv 预印本

配体和氧化剂在 Pd(II) 催化和配体激活的脂肪族羧酸 C(sp 3 )–H 内酯化反应中的作用:机理研究
摘要:研究了 Pd(II) 催化的单 N 保护氨基酸 (MPAA) 配体和 TBHP 氧化剂介导的脂肪族羧酸中 β-C(sp 3 )–H 键内酯化反应的机理。我们已经表明,TBHP 氧化剂和 MPAA 配体的组合非常关键:反应通过 MPAA 配体介导的 TBHP 氧化 Pd(II)/Pd(IV) 进行,然后 Pd(IV) 中间体发生 C–O 还原消除。虽然 Pd(II)/Pd(IV) 氧化是限速步骤,但 C–H 键活化是区域选择性控制步骤。 MPAA 配体不仅可作为辅助配体稳定催化活性物质,还可作为 C–H 键去质子化过程中的质子受体,以及 TBHP 氧化 Pd(II)/Pd(IV) 过程中的质子供体。使用带有羟基的过氧化物基氧化剂也是绝对必要的:在限速 Pd(II)/Pd(IV) 氧化过渡态中,OH 基团的 H 原子参与 1,2-氢转移,以促进 MPAA 配体和过氧化物之间的质子穿梭。因此,脂肪族羧酸中 C(sp 3 )–H 键的内酯化通过 Pd(II)/Pd(IV) 催化循环进行,这与之前报道的 Pd(II) 催化、吡啶酮配体和 O 2 氧化剂辅助的芳香族 o-甲基苯甲酸中苄基 C–H 内酯化不同,后者通过 Pd(II)/Pd(0) 催化循环和分子内 SN 2 亲核取代机理进行。通过比较脂肪族和芳香族羧酸中 C(sp 3 )–H 键内酯化的这些结果,我们能够确定催化剂、底物、配体和氧化剂的作用。

ATP1A1是黑色素瘤治疗的有前途的新靶标,其生理配体Bufalin可以抑制其恢复靶向治疗疗法
摘要尽管在治疗转移性黑色素瘤方面取得了进步,但许多患者对靶向疗法表现出抗性。我们的研究重点是ATP1A1,这是一种与癌症发展相关的钠泵亚基。我们旨在评估黑色素瘤患者的ATP1A1预后价值,并检查其配体Bufalin,体外和体内黑色素瘤细胞系的影响。高ATP1A1表达(IHC)与黑色素瘤患者的总体存活率降低相关。 对BRAF抑制剂的抗性与患者活检(IHC,QPCR)和细胞系(Western blot,QPCR)的ATP1A1水平升高有关。 此外,基于癌症基因组图集(TCGA)数据库和Verfaillie增殖基因签名分析的数据,高的ATP1A1 mRNA表达与分化/色素沉着标记正相关。 bufalin在小窝(接近连接测定法)中特异性靶向ATP1A1,并影响SRC磷酸化(Western blot),从而破坏了多个信号通路(磷酸激酶阵列)。 在体外,Bufalin在ATP1A1(siRNA实验)上作用于ATP1A1(siRNA实验),并在体内使用裸小鼠异种移植模型通过连续的Bufalin通过渗透泵递送,从而诱导黑色素瘤细胞系凋亡。 总而言之,我们的研究表明,ATP1A1可以作为患者生存的预后标志物,也可以作为对BRAF抑制剂治疗的反应的预性标记。 通过靶向ATP1A1,Bufalin抑制细胞增殖,体外诱导凋亡,并有效抑制小鼠的肿瘤发育。高ATP1A1表达(IHC)与黑色素瘤患者的总体存活率降低相关。对BRAF抑制剂的抗性与患者活检(IHC,QPCR)和细胞系(Western blot,QPCR)的ATP1A1水平升高有关。此外,基于癌症基因组图集(TCGA)数据库和Verfaillie增殖基因签名分析的数据,高的ATP1A1 mRNA表达与分化/色素沉着标记正相关。bufalin在小窝(接近连接测定法)中特异性靶向ATP1A1,并影响SRC磷酸化(Western blot),从而破坏了多个信号通路(磷酸激酶阵列)。在体外,Bufalin在ATP1A1(siRNA实验)上作用于ATP1A1(siRNA实验),并在体内使用裸小鼠异种移植模型通过连续的Bufalin通过渗透泵递送,从而诱导黑色素瘤细胞系凋亡。总而言之,我们的研究表明,ATP1A1可以作为患者生存的预后标志物,也可以作为对BRAF抑制剂治疗的反应的预性标记。通过靶向ATP1A1,Bufalin抑制细胞增殖,体外诱导凋亡,并有效抑制小鼠的肿瘤发育。因此,我们的发现强烈支持ATP1A1作为一个有前途的治疗靶标,Bufalin是破坏其肿瘤促进活性的潜在药物。

ATP1A1是黑色素瘤治疗的有前途的新靶标,其生理配体Bufalin可以抑制其恢复靶向治疗疗法
使用三种不同模型的黑色素瘤细胞系(CKIT,BRAF或NRA中的突变)实验。与其敏感的对应物相比,我们的发现始终显示出对靶向疗法的耐药性,分别获得靶向疗法的抗性,分别获得了对靶向疗法的耐药性。在mRNA和蛋白质水平上都观察到了这种增加(图2B,C)。此外,发现在Na+/K+-ATPaseαPUMP的同工型中,ATP1A1在敏感和耐药的MM074和HBL细胞系中均具有最高表达,并比较了四个同工型(ATP1A1-4)(ATP1A1-4)(图2D)。值得注意的是,MM161和MM161-R细胞系也表现出高水平的ATP1A3表达。这些结果提供了进一步的证据

蛋白质-配体氢键强度模式的量子力学评估:来自半经验紧结合和局部振动模式理论的见解
摘要:氢键 (HB) 是生物系统中最丰富的基序。它们在确定蛋白质-配体结合亲和力和选择性方面起着关键作用。我们设计了两个对药物有益的 HB 数据库,数据库 A 包括约 12,000 个蛋白质-配体复合物,约 22,000 个 HB 及其几何形状,数据库 B 包括约 400 个蛋白质-配体复合物,约 2200 个 HB,它们的几何形状和键强度通过我们的局部振动模式分析确定。我们确定了七种主要的 HB 模式,可用作从头 QSAR 模型来预测特定蛋白质-配体复合物的结合亲和力。据报道,甘氨酸是供体和受体谱中最丰富的氨基酸残基,而 N–H · · · O 是数据库 A 中最常见的 HB 类型。HB 倾向于处于线性范围内,且线性 HB 被确定为最强的。HB 角在 100–110° 范围内的 HB 通常形成分子内五元环结构,表现出良好的疏水性和膜通透性。利用数据库 B,我们发现了 2200 多种蛋白质-配体 HB 的广义 Badger 关系。此外,每种氨基酸残基和配体功能团之间的强度和出现图为新颖的药物设计方法和确定药物选择性和亲和力提供了极具吸引力的可能性,它们也可作为命中到先导化合物过程的重要工具。
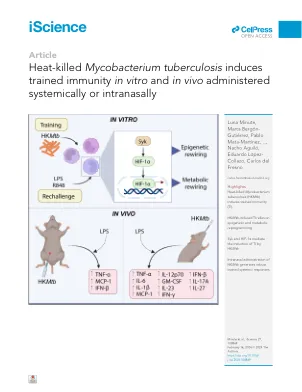
热杀死的结核分枝杆菌诱导受过训练的免疫力Invitro和Invivo在全身或室内施用
摘要训练的免疫(TI)代表了先天免疫细胞的记忆样过程。ti可以用各种化合物(例如真菌B-葡聚糖或结核病疫苗)启动。从未考虑到利用TI防止感染和癌症的临床应用,对新的,简单且易于使用的TI诱导剂的需求日益增长。在这里,我们证明了热杀死的细菌结核(HK MTB)在体外和体内诱导Ti。在人类的单核细胞中,这种效果代表了一个真正受过训练的过程,因为HK MTB赋予针对各种异源挑战的炎症反应,例如脂多糖(Toll-like受体[TLR] 4 Ligand)和R848(TLR7/8 Ligand)(TLR7/8 Ligand)。从机械上讲,HK MTB诱导的Ti依赖于SYK/HIF -1 A依赖性方式的表观遗传机制。在体内,HK MTB在系统地和室内施用时会诱导Ti,而后者产生更健壮的Ti响应。总而言之,我们的研究表明,香港MTB有可能充当粘膜免疫疗法,可以成功诱导训练有素的反应。

结构化的灯光和材料研讨会2024
在基于SESAM的模式模式锁定的半导体激光Yu-Hsin Hsu Hsu(国家Yang-Ming Chiao Tung University)的谐波模式锁定中,谐波模式锁定的动态演变谐波模式锁定的动态演变 and Photoluminescence Property of Gold Clusters with Bis(benzo[b]phosphindole)ethane Ligand Teppei Yahagi (Osaka Metropolitan University) Synthesis and Optical Properties of Gold Nanocluster with Organic Radical Ligand Kosei Hayashi (Osaka Metropolitan University) Numerical investigation of launch characteristics in optical vortex laser induced forward transfer Mamoru Tamura (Osaka University) Helical excitations in superfluid helium Yosuke Minowa (Kyoto University) Fabrication of Hydrogel Fibers with Helical Structure via Vortex Laser Photopolymerization Toward Chiral Tissue Engineering Zhuying Zhang (Osaka University) Development of optical manipulation of nanoscale objects for controlling cellular activity Tatsunori Kishimoto (Toyohashi University技术)的两光子制造微观结构由飞秒光涡流横梁Yoshihisa Matsumoto(大阪大都会大学)谐波模式锁定的动态演变 and Photoluminescence Property of Gold Clusters with Bis(benzo[b]phosphindole)ethane Ligand Teppei Yahagi (Osaka Metropolitan University) Synthesis and Optical Properties of Gold Nanocluster with Organic Radical Ligand Kosei Hayashi (Osaka Metropolitan University) Numerical investigation of launch characteristics in optical vortex laser induced forward transfer Mamoru Tamura (Osaka University) Helical excitations in superfluid helium Yosuke Minowa (Kyoto University) Fabrication of Hydrogel Fibers with Helical Structure via Vortex Laser Photopolymerization Toward Chiral Tissue Engineering Zhuying Zhang (Osaka University) Development of optical manipulation of nanoscale objects for controlling cellular activity Tatsunori Kishimoto (Toyohashi University技术)的两光子制造微观结构由飞秒光涡流横梁Yoshihisa Matsumoto(大阪大都会大学)and Photoluminescence Property of Gold Clusters with Bis(benzo[b]phosphindole)ethane Ligand Teppei Yahagi (Osaka Metropolitan University) Synthesis and Optical Properties of Gold Nanocluster with Organic Radical Ligand Kosei Hayashi (Osaka Metropolitan University) Numerical investigation of launch characteristics in optical vortex laser induced forward transfer Mamoru Tamura (Osaka University) Helical excitations in superfluid helium Yosuke Minowa (Kyoto University) Fabrication of Hydrogel Fibers with Helical Structure via Vortex Laser Photopolymerization Toward Chiral Tissue Engineering Zhuying Zhang (Osaka University) Development of optical manipulation of nanoscale objects for controlling cellular activity Tatsunori Kishimoto (Toyohashi University技术)的两光子制造微观结构由飞秒光涡流横梁Yoshihisa Matsumoto(大阪大都会大学)and Photoluminescence Property of Gold Clusters with Bis(benzo[b]phosphindole)ethane Ligand Teppei Yahagi (Osaka Metropolitan University) Synthesis and Optical Properties of Gold Nanocluster with Organic Radical Ligand Kosei Hayashi (Osaka Metropolitan University) Numerical investigation of launch characteristics in optical vortex laser induced forward transfer Mamoru Tamura (Osaka University) Helical excitations in superfluid helium Yosuke Minowa (Kyoto University) Fabrication of Hydrogel Fibers with Helical Structure via Vortex Laser Photopolymerization Toward Chiral Tissue Engineering Zhuying Zhang (Osaka University) Development of optical manipulation of nanoscale objects for controlling cellular activity Tatsunori Kishimoto (Toyohashi University技术)的两光子制造微观结构由飞秒光涡流横梁Yoshihisa Matsumoto(大阪大都会大学)and Photoluminescence Property of Gold Clusters with Bis(benzo[b]phosphindole)ethane Ligand Teppei Yahagi (Osaka Metropolitan University) Synthesis and Optical Properties of Gold Nanocluster with Organic Radical Ligand Kosei Hayashi (Osaka Metropolitan University) Numerical investigation of launch characteristics in optical vortex laser induced forward transfer Mamoru Tamura (Osaka University) Helical excitations in superfluid helium Yosuke Minowa (Kyoto University) Fabrication of Hydrogel Fibers with Helical Structure via Vortex Laser Photopolymerization Toward Chiral Tissue Engineering Zhuying Zhang (Osaka University) Development of optical manipulation of nanoscale objects for controlling cellular activity Tatsunori Kishimoto (Toyohashi University技术)的两光子制造微观结构由飞秒光涡流横梁Yoshihisa Matsumoto(大阪大都会大学)

Halotag®技术:水溶性配体
图3。使用JaneliaFluor®Halotag®配体的U2OS细胞的活细胞标记表达核Halotag®蛋白。父母U2OS细胞和U2OS细胞稳定地表达与三个核定位序列融合的halotag®蛋白被粘附在玻璃底室载玻片上,并用JaneliaFluor®503,JaneliaFluor®JFX554或Janelia fluor®JFX554或Janeliafluor®635collagang®ligand con Client + 30分钟在30分钟的clienteriafluor®503标记。孵化器。细胞培养基被苯酚红培养基取代。的细胞用488nm激光激发JaneliaFluor®503Halotag®配体(面板A),561nm激光激发JaneliaFluor®JFX554Halotag®Ligand(B)和637nm Laser raser contitation in Janeliafluor®JFX554Halotag®635®635Laseria®6355555。在表达halotag®细胞中,标记仅限于细胞核。父母细胞(无halotag)显示没有标记。使用尼康AX/AXR共聚焦显微镜收集图像,该显微镜具有计划荧光40倍油目标。

细胞因子 - 武装瘤疱疹单纯疱疹病毒:游戏
摘要细胞因子是调节免疫细胞生长和功能活性的小蛋白质,并且已批准进行癌症治疗。溶瘤病毒是通过直接杀死肿瘤细胞并诱导免疫反应来介导抗肿瘤活性的药物。talimogene laherparepvec是一种单纯性疱疹病毒1型(OHSV),批准用于治疗复发性黑色素瘤,该病毒编码人类细胞因子,粒细胞 - 巨噬细胞 - 巨噬细胞刺激性刺激性刺激因子(GM-CSF)。溶瘤病毒的重要优势是能够将治疗有效载荷运送到肿瘤部位,以帮助促进抗肿瘤免疫。虽然细胞因子作为有效载荷特别有趣,但在溶溶病毒中使用的最佳细胞因子仍然存在争议。In this review, we highlight preliminary data with several cytokines and chemokines, including GM-CSF, interleukin 12, FMS- like tyrosine kinase 3 ligand, tumor necrosis factor α , interleukin 2, interleukin 15, interleukin 18, chemokine (C-C motif) ligand 2, chemokine (C-C motif) ligand 5, chemokine (C-X-C序列4或它们的组合,并显示这些有效载荷如何进一步增强OHSV的抗肿瘤免疫力。OHSV对细胞因子递送的更好了解可以帮助改善癌症患者的溶瘤病毒免疫疗法的临床益处。

双重靶向EGFR和MTOR途径通过调节肿瘤微环境来抑制胶质母细胞瘤的生长
摘要:胶质母细胞瘤(GBM)侵袭性生长是由酪氨酸激酶受体(例如表皮生长因子受体(EGFR))的多种信号通路和基因组改变的冗余激活所驱动的,这在50%以上的病例中会改变。靶向EGFR的单个试剂尚未证明对GBM有效。在这项研究中,我们旨在使用对培养和体内的患者衍生的GBM样品进行药物基因组学测试来确定有效的抗肿瘤方案。十个EGFR驱动的GBM样品的高通量药理筛选确定了erlotinib(EGFRI)和MLN0128的组合(雷帕霉素抑制剂或MTORI的哺乳动物靶标)是最有效的抑制肿瘤细胞的可抑制肿瘤细胞的可抑制性肿瘤。Erlonitib+MLN0128的抗肿瘤活性是协同的,并且产生了培养中P-EGFR,有丝分裂原激活的蛋白激酶(MAPK)和磷酸肌醇3-激酶(PI3K)的抑制作用。使用GBM的原位鼠模型,我们表明Erlotinib+MLN0128抑制了体内肿瘤的生长,并且显着延长了肿瘤小鼠的存活率。从经过处理的小鼠中肿瘤组织的表达促进了由Erlotinib+MLN0128诱导的独特基因特征,由肿瘤微环境中免疫抑制趋化因子的下调,包括C-C-C-C-C-C-C-Cotif趋化因子LIGAND LIGAND LIGAND 2(CCL2)和孔孔素。较低的骨膜素水平导致抑制IbA1+(肿瘤促进)巨噬细胞在GBM异种移植物中的效果中。综上所述,我们的结果表明,使用临床可用药物对EGFR和MTOR进行药理学共同定位是EGFR驱动的GBMS的有效治疗范式,既可以通过抑制肿瘤细胞生长并调节免疫性肿瘤微环境。