XiaoMi-AI文件搜索系统
World File Search SystemLumo

支持信息量子振动电子效应...
图 S1:使用 SCAN 函数获得的孤立五金刚烷分子的最低和最高占据分子轨道的模式分辨非谐波测量和电子-声子耦合能量 (EPCE)。上图:根据 100 K 下量子恒温分子动力学模拟获得的轨迹计算出的模式分辨非谐波测量。中图:使用冻结声子方法计算出的最低未占据分子轨道 (LUMO) 的模式分辨 EPCE。下图:使用冻结声子方法计算出的最高占据分子轨道 (HOMO)、HOMO-1 和 HOMO-2 能级的模式分辨 EPCE。

数据驱动的洞察力对锂电池电解质中离子 - 溶剂复合物的还原稳定性的洞察力
摘要:锂(LI)金属电池(LMB)由于其超高理论能量密度而被视为最有前途的储能系统之一。但是,LI阳极的高反应性导致电解质的分解,从而对LMB的实际应用产生了巨大的障碍。常规试验方法在为LI金属阳极设计高度稳定的溶剂分子时效率低下。在此,提出了一种数据驱动的方法来探测溶剂还原稳定性的起源,并加速了晚期电量的分子设计。首先使用基于图理论的算法构建一个潜在溶剂分子的大数据库,然后通过第一原理计算和机器学习(ML)方法进行了全面研究。根据最低无占用分子轨道(LUMO)的分析,在离子 - 溶剂复合物的优势下,99%的电解质的还原稳定性下降。Lumo能级与结合能,键长和轨道比因子有关。基于沙普利添加剂解释的一种可解释的ML方法将偶极矩和分子半径识别为影响协调溶剂的还原性稳定性的最关键描述。这项工作不仅为离子溶剂化学提供了富有成果的数据驱动的见解,而且还揭示了调节溶剂的还原稳定性的关键分子描述子,从而加速了下一代LI Batteries的高级电解质分子的合理设计。8 - 11■简介可充电电池的出现彻底改变了现代技术,催化了大规模网格和无数消费电子产品的开发,例如智能手机,笔记本电脑和电动汽车。1-3,尤其是锂(Li)离子电池(LIBS),是最广泛的可充电电池之一,具有显着改变的能量能量和生活方式习惯的模式。4-7尽管Libs由于明显的优势而占据了可充电电池市场多年的主导地位,但它们的实用能量密度正接近理论上的限制。因此,由于现代社会的需求不断增长,因此需要强烈需要下一代高能密度。
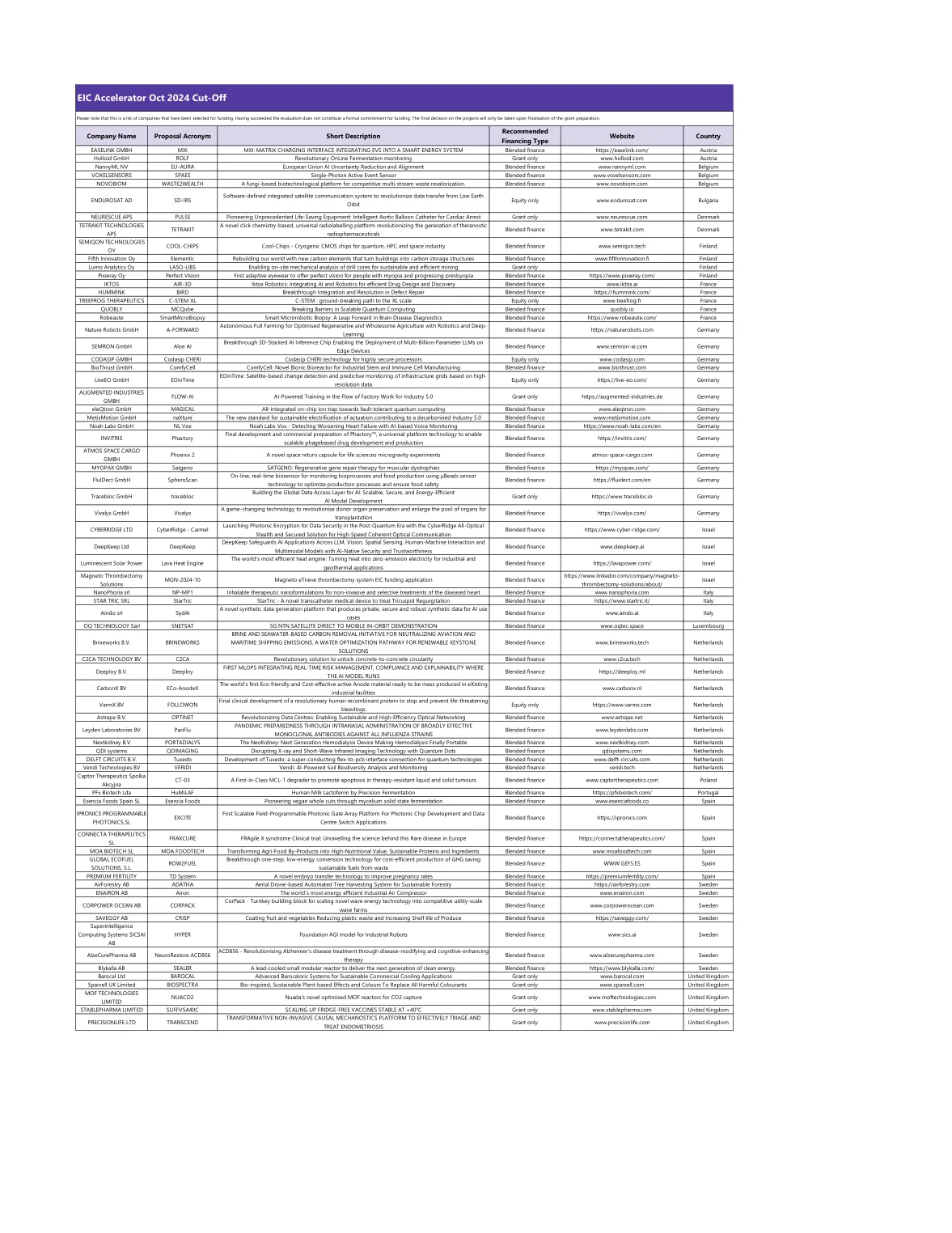
EIC加速器2024年10月 - 欧洲创新委员会
第五创新元素通过新的碳元素重建我们的世界,将建筑物变成碳存储结构,将融资融合www.fifthinnovation.fi Finland Lumo Analytics Oy LASO-LIBS OY LASO-LIBS OY LASO-LIBS实现现场核心,以提供钻井核心,以便为钻机提供完美的型号,以使菲尔德式的迈出适应性的愿景,首先是适应性的,以适用于菲尔斯的幻想,以适用于菲尔斯的幻想。进步的长老会混合融资https://www.pixieray.com/芬兰iktos air-3d iktos机器人技术:整合AI和机器人技术,以进行有效的药物设计和发现混合融合融合www.iktos.iktos.iktos.iktos.iai www.iktos.i C-STEM XL C-STEM:XL量表公平的开创性途径仅www.treefrog.fr法国法国在可伸缩的量子计算中打破障碍的质量障碍,混合融资。第五创新元素通过新的碳元素重建我们的世界,将建筑物变成碳存储结构,将融资融合www.fifthinnovation.fi Finland Lumo Analytics Oy LASO-LIBS OY LASO-LIBS OY LASO-LIBS实现现场核心,以提供钻井核心,以便为钻机提供完美的型号,以使菲尔德式的迈出适应性的愿景,首先是适应性的,以适用于菲尔斯的幻想,以适用于菲尔斯的幻想。进步的长老会混合融资https://www.pixieray.com/芬兰iktos air-3d iktos机器人技术:整合AI和机器人技术,以进行有效的药物设计和发现混合融合融合www.iktos.iktos.iktos.iktos.iai www.iktos.i C-STEM XL C-STEM:XL量表公平的开创性途径仅www.treefrog.fr法国法国在可伸缩的量子计算中打破障碍的质量障碍,混合融资。

1,2,3‐三唑亚基硒的见解
这项工作展示了一段旅程,首先旨在通过对其容易获得的硒加合物进行电化学研究来确定三唑亚甲基的电子性质,然后找到大量还原三唑亚甲基金配合物的光谱证据。此外,我们还报告了通过三唑啉硒酮对自由基阴离子稳定三唑亚甲基过渡金属配合物的 DFT 驱动定向设计。中间站点是硒酮的循环伏安法研究、还原电位与 LUMO 能级的相关性、特定三唑啉硒酮的意外电化学可逆性、还原物种的分析以及从 MIC 硒加合物到过渡金属配合物的电化学性质转移。循环伏安法、EPR 和 UV/Vis 光谱电化学研究、理论计算和合成方法。为了尽最大努力

DNA 碱基的电子附着能
我们发现嘧啶胸腺嘧啶 (T) 和胞嘧啶 (C) 的 VAE 仅相差 0.03 eV,嘌呤鸟嘌呤 (G) 和腺嘌呤 (A) 的 VAE 仅相差 0.08 eV。与“化学”直觉相反,嘧啶的垂直形成的阴离子比较大嘌呤的阴离子更稳定,大约高 0.2 eV。考虑到每种化合物中中性势面和阴离子势面之间的 Franck-Condon 重叠,我们发现所有碱基都有一系列共同的能量,电子可在该能量范围内附着。换句话说,碱基的最低临时阴离子状态在实际意义上是简并的。此外,我们还观察到与腺嘌呤以外所有碱基的最低空分子轨道 (LUMO) 相关的临时阴离子核运动的证据。这表明电子注入这些轨道强烈激发中性分子的振动模式。

金属介导的DNA纳米技术中的3D -CDN
DNA双螺旋含有金属介导的DNA(mMDNA)碱基对由嘧啶:嘧啶对之间的Ag +和Hg 2 +离子构建,并具有纳米电子的承诺。MMDNA纳米材料的合理设计是不切实际的,没有完整的词汇和结构描述。在这里,探索了结构性DNA纳米技术的可编程性,探索了其自我组装的生物分子结构测定平台的自我组装的使命。使用X射线差异构建MMDNA对的全面结构库,并阐明了MMDNA构建的广义设计规则。发现了两种结合模式:N3-主导,中心对称对和由5位环修改驱动的主要凹槽粘合剂。能量差距计算显示了MMDNA结构的最低未居住的分子轨道(LUMO)中的额外水平,使它们具有吸引力的分子电子候选物。

化学教学大纲Nep_syllabus.pdf
Nomenclature for acyclic compounds only (trivial and IUPAC), DBE, hybridization(sp", n= 1,2,3) of C, N, O, halogens, bond distance, bond angles, VSEPR, shapes of molecules, inductive and field effects, bond energy, bond polarity and polarizability, dipole moment, resonance, resonance energy, steric inhibition of resonance,过度结合,𝞹 -M.环,带电的系统3,4,5,7环,融合点,熔点,沸点,氢化热,燃烧热,氢键(内部和分子间),冠 - 酸,酸度的概念,碱性反应中间体:碳定位,碳纤维,自由基,卡宾和硝基的结构和稳定性。

萘化合物的量子计算分析
摘要 这项工作的创新之处在于量子计算分析的应用,具体来说,这项工作采用密度泛函理论 (DFT) 和 Hartree-Fock (HF) 技术以及各种基组 (aug-cc-pVQZ、3-21G、6-31G、6-311G 和 SDD),研究了萘的结构和特性。探索了萘结构和特性的理论性质:最高占据分子轨道 (HOMO)、最低未占据分子轨道 (LUMO)、带隙 BG、态密度 (DOS)、紫外 (UV) 和自然键轨道 (NBO)。研究了几个其他特性:标准温度和压力下的热化学性质及其光学性质(具有间接和直接跃迁的光学 BG)。本研究采用 DFT/aug-cc-pVQZ 基础,以 4.75 eV 为固定值,确定了萘的 HOMO-LUMO 间隙。我们在最近的密度泛函理论 (DFT) 研究中发现间隙分别为 4.71、4.873 和 4.74 eV,与我们的结果一致。

文章_705391_06b199bdb7ee7...
E HOMO ( ( eV) E LUMO ( eV) ΔE 间隙 ( eV) I ( eV) A ( eV) 1 -6.35657 1.210904 7.567469 6.356565 -1.2109 2 -6.0469 1.508324 7.555224 6.046901 -1.50832 3 -6.24609 0.862055 7.108142 6.246087 -0.86205 4 -6.20718 1.361927 7.569102 6.207175 -1.36193 5 -6.38133 0.823687 7.205015 6.381328 -0.82369 6 -6.34677 0.81117 7.157939 6.346769 -0.81117 7 -6.38813 0.926273 7.314404 6.388131 -0.92627 8 -6.54786 0.742869 7.29073 6.547861 -0.74287 9 -6.33507 0.955117 7.290186 6.335068 -0.95512 10 -7.08773 0.620146 7.70788 7.087734 -0.62015 11 -6.36827 1.285191 7.653457 6.368266 -1.28519 12 -7.19086 -0.6011 6.589766 7.190865 0.601098 13 -7.18052 -0.91838 6.262142 7.180524 0.918382 14 -7.10569 -0.74994 6.355749 7.105693 0.749944 15 -7.21508 -0.58287 6.632216 7.215083 0.582867 16 -6.98052 -0.33688 6.643645 6.980521 0.336876 分子的电离势(I)和电子亲和势(A)是量子描述符,

线性共轭四聚体作为表面修饰的层...
表面修饰的层对于钙钛矿太阳能电池的性能和稳定性很重要,但是对表面改性材料的研究仍落后于光伏磁场中的钙钛矿材料。在这项工作中,通过高合成产率的Stille耦合开发了线性共轭的四聚体IDTT4PDI。IDTT4PDI表现出极好的溶解度,热稳定性,合适的Lumo水平(-4.08 eV)和高电子迁移率,这意味着它适合在倒置的钙钛矿太阳能电池中用作表面修饰层。使用IDTT4PDI作为表面修饰的层改善了钙钛矿层和PCBM膜之间的界面接触,减少了陷阱辅助的重组,并提高了电子传输效率。结果,IDTT4PDI-MAPBI 3 PEROVSKITE倒置设备可实现超过20%的效率,该设备远高于控制装置(17%)。这项工作为使用线性二酰亚胺衍生物作为有效的表面修饰材料打开了一个新方向,以实现高效的钙钛矿太阳能电池。