XiaoMi-AI文件搜索系统
World File Search SystemMgCl

nzytaq II 2×无色主混合
nzytaq II 2×无色主混合物是一种预混合的现成溶液,其中含有NZytaq II DNA聚合酶(MB354),属于新一代TAQ衍生的DNA聚合酶,优化了用于标准PCR应用的DNA聚合酶。主混合物含有最佳浓度的DNTP,反应缓冲液和添加剂,并支持最高6 kb的广泛的DNA模板的可靠和可靠放大。MGCL 2最终浓度为2.5 mm,允许实施各种PCR协议。对于高度敏感的下游应用,建议在随后的协议中使用前使用nzygelpure(MB011)净化放大的PCR产物。Nzytaq II DNA聚合酶缺乏3'→5'外切核酸酶活性。由此产生的PCR产品具有A-悬垂性,适用于NZYTECH的NZY-A PCR克隆试剂盒(MB053)或NZY-A-A快速PCR克隆试剂盒(MB137)。

T7 DNA聚合酶 Qiagen验证报告 b'' T7 DNA连接酶 Qiagen®多路复用PCR加套件 T4基因32蛋白
大肠杆菌DNA污染单元测试了N/A N/A 100 100 100规格> 99%13,333 U/mg功能性功能性NO conversion <10份蛋白质来源:重组大肠杆菌菌株,携带毒液T7基因5和E. coli trxa基因。单位定义:1个单位定义为将10 nmol的总DNTPS转换为酸不溶性材料所需的聚合酶量,在37°C下30分钟内。分子量:92.1 KDA质量控制分析:使用2倍连续稀释方法测量单位活动。稀释酶,并将其添加到含有小腿胸腺DNA,1x T7 DNA聚合酶单位表征缓冲液(20 mM Tris-HCl,100 mm KCl,6 mM MGCL,6 mM MGCL 2,6mmmmmgcl 2,0.1 mm EDTA,5 mmβ-MMβ-MERCAPTOETOETHANANOL),3 H-DTT的反应中,3 H-DTT,在37°C下孵育10分钟,浸入冰上,并使用Sambrook和Russell的方法进行分析(6)。蛋白浓度(OD 280)由OD 280吸光度确定。物理纯度,然后进行银色染色检测。通过比较浓缩样品中污染物带的聚集质量与稀释样品中蛋白蛋白蛋白带的质量来评估纯度。单链核酸酶在含有放射性标记的单链DNA底物的50 µL反应中确定,在37°C下孵育4小时4小时。双链外切核酸酶在50 µL反应中确定,该反应含有放射性标记的双链DNA底物和10 µL的酶溶液在37°C下孵育4小时。双链核酸内切酶在50 µL反应中确定,该反应含有0.5 µg质粒DNA和10 µL的酶溶液在37°C下孵育4小时。大肠杆菌16S rDNA的污染是使用5 µL r菌酸溶液的样品变性的样品,并在Taqman QPCR分析中筛选,以使用与16S rRNA locus相应的寡核苷酸引物,使用污染的大肠杆菌Genomic DNA。

确定位置的单键表面的动力学...
我们先前的工作中描述了带有DNA折纸(DNAO)间隔者(DNAO)间隔者(DNAO)隔离剂(DNAO)隔离剂(DNAO)隔离剂(NPOM)构建体(见下文)。[23]简而言之,通过将样品浸入DNAO溶液中,用1-mm MGCL 2,0.5×TBE Buffer浸入DNAO溶液中,用折叠的DNAO模板官能化。aunps用5 0硫醇修饰的20×多-T链功能化,以杂交至少30分钟,而先前折纸先前组装到AU基板上。一旦完全组装,底物用毫克水冲洗并用氮气吹干。AuNP的表面覆盖密度保持足够低,以允许单个AUNP特征。重要的是要注意,溶液中没有凝聚。将所得的干样品放置在配备同时SER的显微镜下,并在单个NP水平上进行深色场表征。sers收集在反向散射的几何形状中,并从0.9-Na,100倍空气放空物镜镜头进行启动。

氮化硼和硬碳
相机械法、液相剥离或液氮中的气体剥离。然而,得到的h-BN材料往往存在表面积低或晶体结构低的问题9-12。最近,我们的研究小组报道了一种使用镁金属将非晶态h-BN转化为结晶h-BN的策略。13然而,这种熔融金属熔剂方法需要严格的转变条件(900℃),并且即使在热处理后采用酸洗程序也会引入潜在的杂质。此外,液态镁金属易燃,需要严格的惰性气体条件以及独特的不锈钢高压釜。另外,金属熔剂法不能控制反应并实现所需的结晶程度。在此,我们报道了一种优越的电化学方法,避免了使用熔融镁金属及其相关的安全隐患。我们假设是否有可能利用熔融的 MgCl 2 原位生成 Mg 金属,类似于之前使用熔融的 CaCl 2 的过程。14, 15

使用合成的激子传输工程耦合...
图 1 | 使用 DNA 支架形成 Cy3 聚集体的化学方法。 (a) Cy3 (左) 共价连接到单链 DNA (ss-DNA) 脱氧核糖磷酸骨架的 3' 和 5' 端。 Cy3 修饰的 DNA 纳米结构是通过将 Cy3 修饰的 ssDNA 与规范互补的 ssDNA 链杂交而形成的,如连接到 DNA 双链体的 Cy3 单体的分子动力学快照 (中间) 和示意图 (右、上) 中蓝色椭圆表示 Cy3 所示。 Cy3 二聚体和三聚体是通过将连续的 Cy3 发色团连接到 ssDNA 并与互补链杂交而形成的 (右、中和下) (b) Cy3 单体 (棕色)、二聚体 (蓝色) 和三聚体 (绿色) 的吸光度 (实线) 和量子产率归一化的荧光光谱 (虚线)。 [DNA 双链] = 0.5 µ M,溶于 40 mM Tris、20 mM 醋酸盐、2 mM 乙二胺四羧酸 (EDTA) 和 12 mM MgCl 2 (TAE-MgCl 2 缓冲液)。(c) 双链中 Cy3 单体、二聚体和三聚体的荧光量子产量 (ΦF)。[DNA 双链] = 0.5 µ M,溶于 1 × TAE-MgCl 2 缓冲液。(d) Cy3 单体、二聚体和三聚体的圆二色性 (CD) 光谱。(e) Cy3 单体、二聚体和三聚体的荧光衰减轨迹,仪器响应函数以黑色显示。

li – mg – zr – cl尖晶石
摘要:基于氯化物的固体电解质是由于其高LI +离子电导率和与高压氧化物阴极的全溶剂锂电池相关的材料而引人入胜的材料。然而,这些材料的主要示例仅限于三价金属(例如SC,Y和IN),这些金属价格昂贵且稀缺。在这里,我们通过用二二元和四价金属(例如Mg 2+和Zr 4+)代替三价金属来扩展这种材料家族。我们合成李2 mg 1/3 zr 1/3 cl 4在尖晶石晶体结构中,并将其性质与先前报道的高性能LI 2 SC 2/3 Cl 4进行比较。我们发现Li 2 mg 1/3 Zr 1/3 cl 4的离子电导率较低(在30°C时为0.028 ms/cm),比同构结构LI 2 SC 2/3 Cl 4(30°C时1.6 ms/cm)。我们将这种差异归因于Mg 2+和Zr 4+在LI 2 mg 1/3 Zr 1/3 Cl 4中的无序排列,这可能会阻止LI+迁移途径。但是,我们表明,Li 2 -Z Mg 1 - 3 Z /2 Zr Z Cl 4之间的Aliovalent取代在Li 2 MgCl 4和Li 2 Zrcl 6之间可以提高离子电导率,而ZR 4+含量的增加,可能是由于引入了Li +空位。这项工作为基于卤化物的固体电解质打开了一个新的维度,从而加快了低成本固态电池的开发。■简介
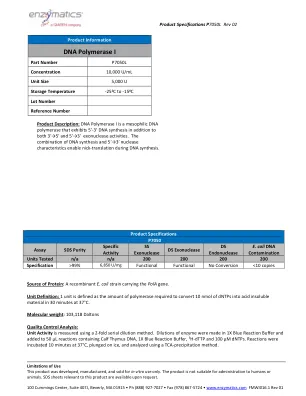
DNA聚合酶I
蛋白浓度(OD 280)由OD 280吸光度确定。物理纯度,然后进行银色染色检测。通过比较浓缩样品中污染物带的聚集质量与稀释样品中蛋白蛋白蛋白带的质量来评估纯度。单链核酸酶在含有放射性标记的单链DNA底物的50 µL反应中确定,在37°C下孵育4小时4小时。双链外切核酸酶在50 µL反应中确定,该反应含有放射性标记的双链DNA底物和10 µL的酶溶液在37°C下孵育4小时。双链核酸内切酶在50 µL反应中确定,该反应含有0.5 µg质粒DNA和10 µL的酶溶液在37°C下孵育4小时。e.coli 16S rDNA污染使用5 µL R含量的酶溶液的样品变性并在Taqman QPCR测定中筛选,以使用污染的大肠杆菌基因组DNA,使用寡核苷酸引物污染了与16S rRNA locus相对应的寡核苷酸引物。提供:25mm Tris-HCl,1mm DTT,0.1mm EDTA,50%甘油(25°C时pH 7.4)。提供:10倍蓝色缓冲区(B0110):500mm NaCl,100mm Tris-HCl,100mm MGCL 2,10mm DTT(25 c)pH 7.9 pH 7.9)。用法说明:5´-overhang(1)

堆叠和配位对于 Ti3C2T2 MXene 中 Li、Na 和 Mg 扩散和插入的重要性
Cheng 等人 [1] 实现了与 Li 0.7 Ti 3 C 2 T 2 相当的容量(1C 电流密度下经过 200 次循环后容量为 100 mAh g −1,电流密度约为 100 mA g −1),而 Wang 等人 [2] 实现了与 ≈ Na 0.5 Ti 3 C 2 T 2 相当的容量(200 mA g −1 电流密度下经过 1000 次循环后容量为 70 mAh g −1,电流密度约为 3C)。隧道电子显微镜(TEM)还显示,在某些情况下可以插入多层 Na,[2] 在原子水平上每个原子级分子式单位可以有一个以上的 Na,即 Na > 1 Ti 3 C 2 T x 。另一方面,Mg 是一种在电池应用中具有挑战性的金属,其扩散速度慢、电解质-电极动力学复杂、质子嵌入和电解质分解问题严重[5–7],在微米级 Ti 3 C 2 T x 上测试时,仅显示出与 Mg 0.004 Ti 3 C 2 T 2 (≈ 1 mAh g − 1,25 次循环) 相当的容量。[3] 使用间隔基增加层间距离 [8] 和/或将 MXene 纳米化 [9,10] 已显示出更高的容量,但很难确定这些容量是由于可逆的 Mg 2 + 嵌入,还是由于表面反应、质子嵌入和/或电解质共嵌入 (如 MgCl + 嵌入的情况)。[5,6,11]

TAQ-B DNA聚合酶
蛋白质的来源:一种重组大肠杆菌菌株,携带来自嗜热有机体Thermus aquaticus YT-1的TAQ DNA聚合酶基因。单位定义:1个单位定义为将在75°C的30分钟内将10 nmol的DNTP纳入酸 - 不溶性材料的酶。分子量:93,910 Daltons质量控制分析:使用2倍连续稀释方法测量单位活动。在1X反应缓冲液中制成酶的稀释液,并将其添加到含有小腿胸腺DNA,25 mM TAPS(pH 9.3),50 mM KCl,2.0mm MGCL2,1 mM DTT,3H-DTTP和100 µm DNTP的50 µL反应中。 在75°C下孵育10分钟,浸入冰上,并使用Sambrook和Russell的方法进行分析(Molecular Cloning,V3,2001,pp。 A8.25-A8.26)。 蛋白浓度(OD 280)由OD 280吸光度确定。 通过浓缩和稀释酶溶液的SDS-PAGE评估物理纯度,然后进行银色染色检测。 通过比较浓缩样品中污染物带的聚集质量与稀释样品中蛋白蛋白蛋白带的质量来评估纯度。 单链核酸酶在含有放射性标记的单链DNA底物的50 µL反应中确定,在37°C下孵育4小时4小时。 双链外切核酸酶在50 µL反应中确定,该反应含有放射性标记的双链DNA底物和10 µL的酶溶液在37°C下孵育4小时。 双链核酸内切酶在50 µL反应中确定,该反应含有0.5 µg质粒DNA和10 µL的酶溶液在37°C下孵育4小时。在1X反应缓冲液中制成酶的稀释液,并将其添加到含有小腿胸腺DNA,25 mM TAPS(pH 9.3),50 mM KCl,2.0mm MGCL2,1 mM DTT,3H-DTTP和100 µm DNTP的50 µL反应中。在75°C下孵育10分钟,浸入冰上,并使用Sambrook和Russell的方法进行分析(Molecular Cloning,V3,2001,pp。A8.25-A8.26)。蛋白浓度(OD 280)由OD 280吸光度确定。物理纯度,然后进行银色染色检测。通过比较浓缩样品中污染物带的聚集质量与稀释样品中蛋白蛋白蛋白带的质量来评估纯度。单链核酸酶在含有放射性标记的单链DNA底物的50 µL反应中确定,在37°C下孵育4小时4小时。双链外切核酸酶在50 µL反应中确定,该反应含有放射性标记的双链DNA底物和10 µL的酶溶液在37°C下孵育4小时。双链核酸内切酶在50 µL反应中确定,该反应含有0.5 µg质粒DNA和10 µL的酶溶液在37°C下孵育4小时。

原发性代谢调整的自然变化决定了模型中的铵耐性。
酶活性通过用500μl的提取缓冲液进行vig口摇(20%(v/v)甘油,1%triton X-100(v/v),50 mm hepes – koH(ph 7.5),10 mm mgcl 2,1 mm edta,1%triton x-100(v/v),1%Triton X-100(V/V),1%Triton X-100(V/V),1%Triton X-100(V/V),1%Triton X-100(V/V),1%Triton X-100(V/V),1%Triton X-100(V/V),1%Triton X-100(v/v),1%X-100(v/v),1%MM emMM MM E.酸,1 mm苯甲米丁,20μM亮肽素,0.5 mM DTT,1 mM苯基甲基磺酰基氟化物,10%聚乙烯基 - 丙吡咯烷酮(W/V)]。葡萄糖激酶(GK),FRUC TOKINAPE(FK),谷氨酸脱氢酶(GDH),磷酸烯醇丙酮酸羧化酶(PEPC),苹果酸脱氢酶(MDH),丙酮酸激酶(PK),总浓酸酯(CM),米尔酸酯(CS),米尔酸酯(CM),米尔酸酯(CM)通过分光光度法测定NADP依赖性的异戊酸脱氢酶(ICDH)酶,并用机器化的微孔板测定法测定(Gibon等人。,2004)。在25°C孵育后,NAD(P)H的演变在340 nm处被固定在340 nm处。通过循环反应在570 nm处测量了GDH的活性,涉及在存在醇脱氢酶和苯嗪硫代硫酸盐的情况下,涉及3-(4,5-二甲基噻唑-2-基)-2,5-二苯基四唑四唑。cs ac ac titive。(2003)。通过检查生物标准(番茄叶提取物)的恢复,并确保提取物的稀释对活动的估计没有影响,如Bénard和Gibon(2016)所述,可以通过检查生物标准的恢复(番茄叶提取物)来验证。