XiaoMi-AI文件搜索系统
World File Search SystemNRAS

应对数字资产非法融资风险行动计划
该行动计划响应了第 14067 号行政命令 (EO) 第 7(c) 节“确保负责任地开发数字资产”的规定,该规定要求制定一项协调的跨机构行动计划,以减轻美国政府《打击恐怖主义和其他非法融资的国家战略》(《非法融资战略》)中确定的数字资产相关的非法融资和国家安全风险。1《非法融资战略》由美国财政部的国家风险评估 (NRA) 2 提供信息,概述了优先事项和支持行动,以确保美国政府使我们的反洗钱/打击恐怖主义融资 (AML/CFT) 制度适应不断变化的威胁环境,并考虑到金融服务和市场的结构性和技术变化。

PAN -RAF抑制剂Tovorafenib患者的1阶段研究...
BRAF和NRA的抽象目的基因组改变是恶性黑色素瘤和其他实体瘤中的致癌驱动因素。Tovorafenib是一种研究,口服,选择性,CNS-PENETRANT,小分子,II型PAN-RAF抑制剂。这项第一个人类1期研究探讨了Tovorafenib的安全性和抗肿瘤活性。方法对复发或难治性晚期实体瘤的成年患者进行了两部分研究,包括剂量升级阶段和剂量扩张阶段,包括分子定义的黑色素瘤患者。主要目标是每隔一天(Q2D)或每周一次(QW)评估一次Tovorafenib的安全性,并在这些时间表上确定最大耐受性和建议的2阶段剂量(RP2D)。次要目标包括评估抗肿瘤活性和Tovorafenib药代动力学。对149例患者进行了tovorafenib的结果(Q2d n = 110,QW n = 39)。Tovorafenib的RP2D定义为200 mg Q2d或600 mg QW。在剂量扩张阶段,Q2D队列中的80名患者中有58例(73%),QW队列中的19名患者中有9名(47%)发生≥3级不良事件。这些总体中最常见的是贫血(14例,14%)和丘疹性皮疹(8例患者,8%)。在Q2D扩张阶段的68名可评估患者中,有10例(15%)在16例(50%)BRAF突变阳性黑色素瘤中的16例患者中有10例(15%)的反应,其中包括RAF和MEK抑制剂。在QW剂量扩张阶段,NRAS突变阳性黑色素瘤对RAF和MEK抑制剂的可评估患者没有反应。 9名患者(53%)对稳定疾病的反应最佳。QW剂量给药与Tovorafenib在400-800 mg的全身循环中的最小积累有关。结论两种时间表的安全性均可接受,QW以600 mg QW的RP2D剂量为将来的临床研究首选。Tovorafenib在BRAF突变的黑色素瘤中的抗肿瘤活性是有希望的,并且在多种环境中持续临床发育是合理的。clinicaltrials.gov标识符NCT01425008。

癌症16-00091-1.pdf
简单的摘要:组蛋白脱乙酰基酶(HDAC)抑制剂是抗癌药,在血液学恶性肿瘤中具有效率;但是,由于治疗指数狭窄和次优地型选择性,可用代理的实用性受到限制。Bocodepsin(OKI-179)是一种新颖的,可生物可利用的I类靶向Depsipeptide HDAC HDAC抑制剂,在临床前实体瘤模型中具有有希望的抗癌活性。在本人类的第一阶段临床研究中,我们报告了按间歇性和连续给药时间表进行口服口服的OKI-179的安全性和耐受性。OKI-179的高度不良事件发生率良好,这支持了OKI-179与其他靶向抗癌剂成功组合的潜力。OKI-179目前正在与MEK抑制剂Binimetinib结合使用NRAS突变的黑色素瘤患者。

RAS 和 BRAF 基因作为个性化结直肠癌治疗的生物标志物和靶点:最新进展
结直肠癌 (CRC) 仍然是最常见的癌症之一,并且发病率一直呈上升趋势。它也是全球最致命的疾病之一,是癌症相关死亡的第三大原因。根据分期和疾病状况,CRC 可通过手术、化疗、放疗、免疫疗法或联合疗法治疗。然而,这些疗法的效果不佳且有不良副作用,因此人们正在不断努力探索新型治疗方式。在 CRC 的亚型中,KRAS、BRAF 和 NRAS 突变的 CRC 分别占总病例的约 43%、10% 和 3%。这些突变与肿瘤进展和抗表皮生长因子受体 (EGFR) 治疗耐药性有关。由于它们在 CRC 中的重要作用,这些基因已成为开发新疗法的靶点。在本文中,我们讨论了针对这些突变基因的 CRC 的当前和未来治疗方法,由于 CRC 中突变发生率较高,我们将更多地关注 KRAS 和 BRAF。


改善
The ICARUS project will produce results that progress the current State of the Art and will be of immediate benefit to Road Authorities: • An overview of the baseline for climate change resilience assessments, resilience evaluation and the use of cost benefit assessments for climate change adaptation • A report on using impact chains to better understand direct and indirect impacts • Guidelines on how to define and use minimum viable service levels for evaluating resilience and adaptation options based关于量化和估值的协会成本和更高的收益,并考虑整个生命的角度•提供概述和措施表征的指南,旨在实施各种NRAS流程的实施(计划,计划,设计,构建,维护,维护,预测)•演示•展示适应性培训如何在基于自然培训方面的范围••针对自然培训的范围•••对实施自然培训的范围,•
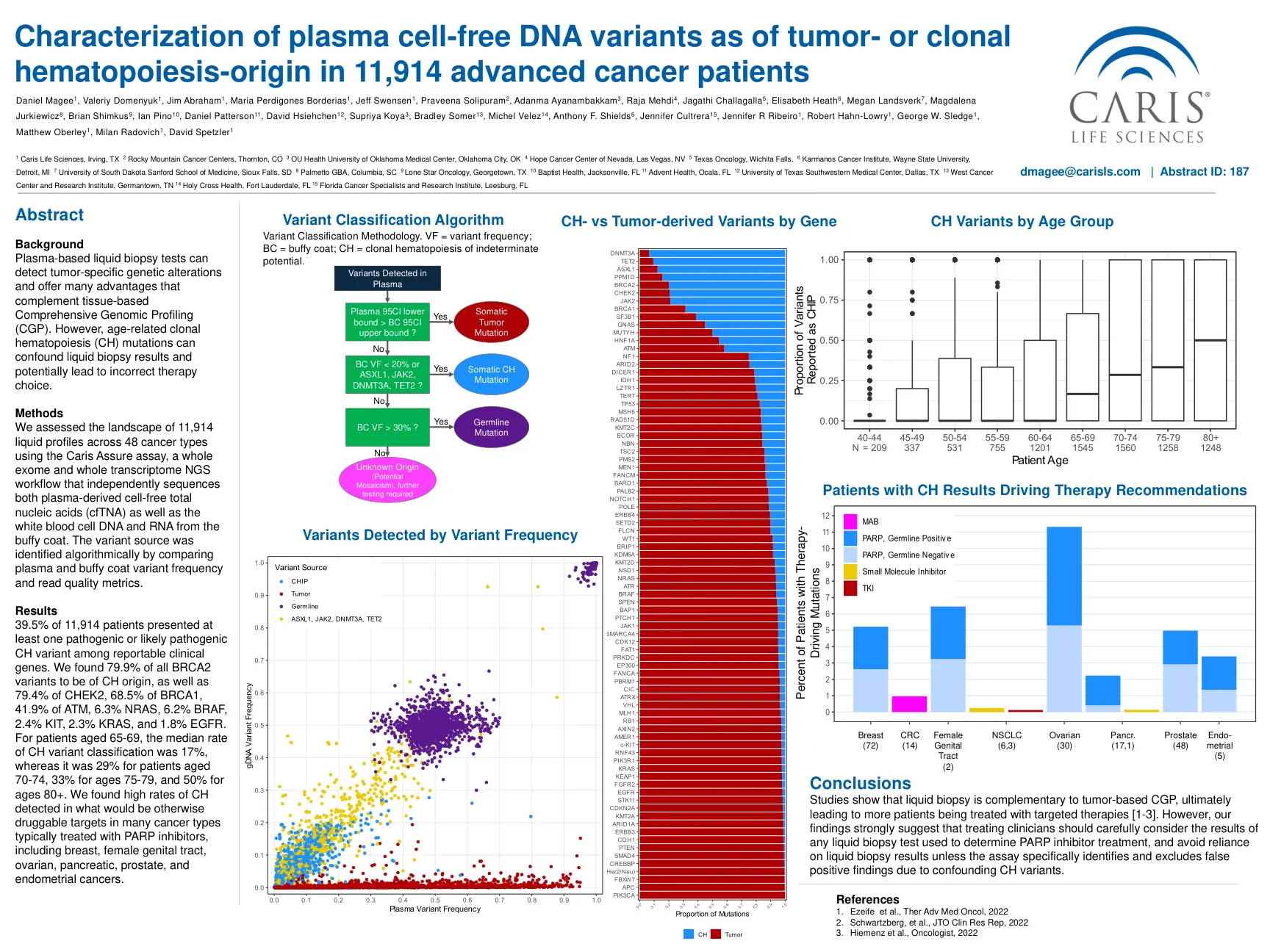
表征无血浆细胞的DNA变体与肿瘤的表征
结果中有39.5%的114例患者在可报告的临床基因中至少提出了一种致病性或可能的致病性CH变体。我们发现所有BRCA2变体的79.9%是CH起源的,占CHEK2的79.4%,BRCA1的68.5%,41.9%的ATM,6.3%NRA,6.2%BRAF,2.4%KIT,2.3%KRAS,2.3%KRAS和1.8%的EGFR。对于65-69岁的患者,CH变体分类的中位数率为17%,而70-74岁的患者为29%,75-79岁的患者为33%,80岁以上的患者为50%。我们发现,在许多通常用PARP抑制剂治疗的癌症类型中,否则可吸毒靶标的CH速率很高,包括乳腺癌,女性生殖道,卵巢,胰腺,胰腺,前列腺和子宫内膜癌。

肿瘤学临床途径 - 急性髓细胞性白血病(AML)
a Diagnosis must include flow cytometry, karyotype, rapid order (<72 hours) molecular tests (to include: FLT3, NPM1, IDH1, and IDH2), and myeloid NGS test (at minimum must include: ASXL1, BCOR, CEBPA, EZH2, FLT3, IDH1, IDH2, NPM1, RUNX1, SF3B1, srsf2,stag2,tp53,u2af1和zrsr2);其他可选基因包括:CBL,DDX41,KIT,KRAS,NRA和其他与髓样肿瘤相关的基因;也可以在病理学家的前提下或自行决定进行AML鱼类测试(可以包括:-5/5Q,-7/7Q,-7/7Q,KMT2A,T(8; 21)runx1 :: Runx1t1,t(15; 17; 17)PML :: Rara,t(16:16)或Inv(16:16)或Inv(16)或Inv(16)CBFB :: bbfb :: bbl :: bbl:n; TP53)

泛 RAF 抑制剂托沃拉非尼的 1 期研究......
摘要目的 BRAF 和 NRAS 的基因组变异是恶性黑色素瘤和其他实体瘤的致癌驱动因素。托沃拉非尼是一种在研的口服、选择性、中枢神经系统渗透性、小分子 II 型泛 RAF 抑制剂。这项首次用于人体的 1 期研究探讨了托沃拉非尼的安全性和抗肿瘤活性。方法这项针对复发或难治性晚期实体瘤成年患者的两部分研究包括剂量递增期和剂量扩展期,包括分子定义的黑色素瘤患者群。主要目标是评估每隔一天 (Q2D) 或每周 (QW) 一次给药的托沃拉非尼的安全性,并确定这些方案的最大耐受剂量和推荐的 2 期剂量 (RP2D)。次要目标包括评估抗肿瘤活性和托沃拉非尼药代动力学。结果 149 名患者(Q2D n = 110,QW n = 39)接受了托沃拉非尼治疗。托沃拉非尼的 RP2D 定义为 200 mg Q2D 或 600 mg QW。在剂量扩展阶段,Q2D 队列中的 80 名患者中有 58 名(73%)和 QW 队列中的 19 名患者中有 9 名(47%)出现 ≥ 3 级不良事件。总体而言,最常见的不良事件是贫血(14 名患者,14%)和斑丘疹(8 名患者,8%)。在 Q2D 扩展阶段,68 名可评估患者中有 10 名(15%)出现反应,包括 16 名(50%)未使用过 RAF 和 MEK 抑制剂的 BRAF 突变阳性黑色素瘤患者中的 8 名。在 QW 剂量扩展阶段,17 名可评估的 NRAS 突变阳性黑色素瘤患者未接受过 RAF 和 MEK 抑制剂治疗,未出现反应;9 名患者 (53%) 的最佳反应为病情稳定。400-800 毫克剂量范围内,QW 剂量给药与体循环中托沃拉非尼的最小蓄积相关。结论两种方案的安全性均可接受,未来临床研究首选 RP2D 600 毫克 QW 剂量。托沃拉非尼在 BRAF 突变黑色素瘤中的抗肿瘤活性很有希望,值得在多种环境中继续进行临床开发。ClinicalTrials.gov 标识符 NCT01425008。

PAN -RAF抑制剂Tovorafenib在...
BRAF和NRA的抽象目的基因组改变是恶性黑色素瘤和其他实体瘤中的致癌驱动因素。Tovorafenib是一种研究,口服,选择性,CNS-PENETRANT,小分子,II型PAN-RAF抑制剂。这项第一个人类1期研究探讨了Tovorafenib的安全性和抗肿瘤活性。方法对复发或难治性晚期实体瘤的成年患者进行了两部分研究,包括剂量升级阶段和剂量扩张阶段,包括分子定义的黑色素瘤患者。主要目标是每隔一天(Q2D)或每周一次(QW)评估一次Tovorafenib的安全性,并在这些时间表上确定最大耐受性和建议的2阶段剂量(RP2D)。次要目标包括评估抗肿瘤活性和Tovorafenib药代动力学。对149例患者进行了tovorafenib的结果(Q2d n = 110,QW n = 39)。Tovorafenib的RP2D定义为200 mg Q2d或600 mg QW。在剂量扩张阶段,Q2D队列中的80名患者中有58例(73%),QW队列中的19名患者中有9名(47%)发生≥3级不良事件。这些总体中最常见的是贫血(14例,14%)和丘疹性皮疹(8例患者,8%)。在Q2D扩张阶段的68名可评估患者中,有10例(15%)在16例(50%)BRAF突变阳性黑色素瘤中的16例患者中有10例(15%)的反应,其中包括RAF和MEK抑制剂。在QW剂量扩张阶段,NRAS突变阳性黑色素瘤对RAF和MEK抑制剂的可评估患者没有反应。 9名患者(53%)对稳定疾病的反应最佳。QW剂量给药与Tovorafenib在400-800 mg的全身循环中的最小积累有关。结论两种时间表的安全性均可接受,QW以600 mg QW的RP2D剂量为将来的临床研究首选。Tovorafenib在BRAF突变的黑色素瘤中的抗肿瘤活性是有希望的,并且在多种环境中持续临床发育是合理的。clinicaltrials.gov标识符NCT01425008。