XiaoMi-AI文件搜索系统
World File Search SystemNusinersen

对脊髓肌萎缩症患者体重≤17kg且≤24个月大的患者的骨nasegene abeparvovec的安全性和耐受性:
关键纳入标准•基于基因突变分析的症状SMA诊断,双重SMN1突变(缺失或点突变)和任何数量SMN2基因的副本•治疗时的年龄≤24个月•重量≤17kg筛选时访问时访问或批准的药物均可进行批准的药物,•纽约批准的药物/牢固的纽约,键/牢记的键abeparvovec在筛查前的四个星期内使用或先前使用任何AAV9基因疗法•抽吸史或吸气迹象(例如,食物的咳嗽或溅射)在筛查前的四个星期内在筛选前15天内

1 Musclesense -UCL Discovery
1 Deenen,J。C. W.等。神经肌肉疾病的流行病学:文献的全面概述。J. Neuromuscul。dis。2,73–85(2015)。 2 Finkel,R。S.等。 Nusinersen与婴儿性脊柱肌肉萎缩中的假手术对照。 N. Engl。 J. Med。 377,1723–1732(2017)。 3 Mendell,J。R.等。 单剂量基因替代疗法用于脊柱肌肉萎缩。 N. Engl。 J. Med。 377,1713–1722(2017)。 4 Voit,T。等。 Drisapersen对Duchenne肌肉营养不良的治疗的安全性和功效(需求II):探索性,随机,安慰剂对照的2期研究。 柳叶刀神经。 13,987–996(2014)。 5临床trials.gov。 可用:https://clinicaltrials.gov/。 (访问:2019年9月7日)2,73–85(2015)。2 Finkel,R。S.等。Nusinersen与婴儿性脊柱肌肉萎缩中的假手术对照。N. Engl。J. Med。 377,1723–1732(2017)。 3 Mendell,J。R.等。 单剂量基因替代疗法用于脊柱肌肉萎缩。 N. Engl。 J. Med。 377,1713–1722(2017)。 4 Voit,T。等。 Drisapersen对Duchenne肌肉营养不良的治疗的安全性和功效(需求II):探索性,随机,安慰剂对照的2期研究。 柳叶刀神经。 13,987–996(2014)。 5临床trials.gov。 可用:https://clinicaltrials.gov/。 (访问:2019年9月7日)J. Med。377,1723–1732(2017)。3 Mendell,J。R.等。 单剂量基因替代疗法用于脊柱肌肉萎缩。 N. Engl。 J. Med。 377,1713–1722(2017)。 4 Voit,T。等。 Drisapersen对Duchenne肌肉营养不良的治疗的安全性和功效(需求II):探索性,随机,安慰剂对照的2期研究。 柳叶刀神经。 13,987–996(2014)。 5临床trials.gov。 可用:https://clinicaltrials.gov/。 (访问:2019年9月7日)3 Mendell,J。R.等。单剂量基因替代疗法用于脊柱肌肉萎缩。N. Engl。J. Med。 377,1713–1722(2017)。 4 Voit,T。等。 Drisapersen对Duchenne肌肉营养不良的治疗的安全性和功效(需求II):探索性,随机,安慰剂对照的2期研究。 柳叶刀神经。 13,987–996(2014)。 5临床trials.gov。 可用:https://clinicaltrials.gov/。 (访问:2019年9月7日)J. Med。377,1713–1722(2017)。4 Voit,T。等。Drisapersen对Duchenne肌肉营养不良的治疗的安全性和功效(需求II):探索性,随机,安慰剂对照的2期研究。柳叶刀神经。13,987–996(2014)。 5临床trials.gov。 可用:https://clinicaltrials.gov/。 (访问:2019年9月7日)13,987–996(2014)。5临床trials.gov。可用:https://clinicaltrials.gov/。(访问:2019年9月7日)

特异性,协同性和剪接修改药物的机制
Title: Specificity, synergy, and mechanisms of splice-modifying drugs Authors : Yuma Ishigami 1,† , Mandy S. Wong 1,2,† , Carlos Martí-Gómez 1 , Andalus Ayaz 1 , Mahdi Kooshkbaghi 1 , Sonya Hanson 3 , David M. McCandlish 1 , Adrian R. Krainer 1,* , Justin B. Kinney 1,*。隶属关系:1。Cold Spring Harbour实验室,纽约州冷泉港,美国11724,美国。2。当前地址:横梁治疗学,马萨诸塞州剑桥,美国02142,美国。3。flatiron Institute,纽约,纽约,10010,美国。†同等贡献。*通信:krainer@cshl.edu(ark),jkinney@cshl.edu(jbk)。摘要:针对MRNA剪接的药物具有很大的治疗潜力,但是对这些药物的工作原理的定量了解受到限制。在这里,我们引入了机械解释的定量模型,以针对剪接修改药物的序列特异性和浓度依赖性行为。使用大量平行的剪接测定,RNA-seq实验和精确剂量反应曲线,我们获得了两种用于治疗脊柱肌萎缩的两种小分子药物Risdiplam和Branaplam的定量模型。的结果定量地表征了Risdiplam和Branaplam对于5'剪接位点序列的特异性,这表明Branaplam通过两种不同的相互作用模式识别5'剪接位点,并证明了SMN2 Exon 7的Risdiplam活性的普遍的两点假设。结果还表明,在小分子药物和反义寡核苷酸药物中,异常的单药合作以及多药协同作用是促进外生包容的。Nusinersen 11–我们的定量模型阐明了现有治疗的机制,并为新疗法的合理发展提供了基础。引言替代性mRNA剪接已成为药物发育的主要重点1-10。已经开发了三种剪接改良药物 - Nusinersen,Risdiplam和Branaplam,以治疗脊柱肌肉萎缩(尽管Branaplam已撤回)。所有三种药物都通过促进SMN2外显子7。

GT Advisory_中国在行动:2021年中国全国药品价格谈判的经验教训
据悉,国家医保局共收到501份申请,涉及474个药品,271个药品通过了初审。国家医保局网站公布了这271个药品的分类,披露了通用名、上市许可持有人名称、适应症、是否存在专利纠纷、使用剂量、有效性和安全性描述等信息。然而,2021年11月初的谈判中,只有117个药品被纳入。复星凯特的CAR-T产品Yescarta(axicabtagene ciloleucel)每针约120万元,虽然被纳入了候选名单,但并未进入谈判。相比之下,百健的Spinraza(通用名:nusinersen)治疗罕见病脊髓性肌萎缩症(SMA),单针约70万元,尽管过去曾失败,但最终被纳入2021年国家医保目录。
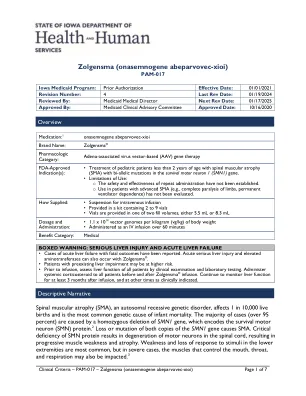
Zolgensma(onasengene abeparvovec-xioi)
结合SMN1的损失,患者保留了第二个相似基因SMN2的可变数量,这些副本可产生降低的生存运动神经元(SMN)蛋白水平,而这对于正常运动神经元功能不足。4 SMN2的副本数量较高,通常与温和的疾病相关,但是这种相关性是相对的,而不是绝对的相关性。5Zolgensma®(Onasengene Abeparvovec-XIOI)是一种基于AAV9的重组基因疗法,旨在提供编码人类SMN蛋白的基因的副本。在人类案例研究中,静脉内(IV)Zolgensma®的给药导致SMN蛋白的细胞转导和表达。脊柱肌肉萎缩已根据症状严重程度和基因型分类为0-4,但是有了新的疗法(包括Nusinersen,Risdiplam和Onasemnogene abeparvovec-Xioi),表格已经变得更加多样化和分类以集中在功能或治疗响应上。脊柱肌肉萎缩的分类6

Swapnil Tirmanwar,Int。 J. of Pharm。 Sci。,2025,第3卷,问题01,131-136
脊柱肌肉萎缩(SMA)是一种主要影响运动神经元的遗传神经肌肉疾病,导致渐进的肌肉无力和萎缩。它是由SMN1基因突变引起的,这导致生存运动神经元(SMN)蛋白的水平降低,这对于肌肉功能必不可少。SMA以不同类型的形式呈现,从严重的婴儿发作形式到较轻的成人发作病例,以及影响流动性,呼吸和运动发育的症状。此临床评论概述了SMA的遗传基础,分类,症状,症状,症状和诊断方法。它还研究了治疗策略的进步,包括基因疗法(例如Onasemnogene Abeparvovec),SMN2剪接修饰剂(如Nusinersen)以及支持护理以改善生活质量。该评论进一步强调了早期诊断,疗法的可及性以及新生儿筛查计划对更好结果的重要性。目标是对SMA进行全面的了解,以告知临床医生,看护人和公共卫生从业人员有效的疾病管理和新兴治疗选择。

三个SMN2基因副本对脊柱肌肉萎缩患者的临床特征和疾病改良治疗的影响的影响:系统文献综述
结果:我们的搜索确定了44项研究,研究了三个SMN2副本对临床特征的影响(21在表型上,自然历史上的13,功能状态和其他体征/症状)。在患有SMN1缺失的I型SMA或预症状的婴儿中,与两份SMN2副本相比,三个SMN2副本与后来的症状发作,运动功能较慢和更长的存活率相关。在患有II型SMA或III型患者中,与四个SMN2副本相比,三个SMN2副本与早期症状发作,移动丧失和呼吸机依赖性有关。11项研究检查了Nusinersen的治疗效果(9项研究),Onasemnogene Abeparvovec(一项研究),以及三种SMN2副本患者的一系列治疗(一项研究)。在预症状的婴儿中,早期治疗延迟了症状的发作,并在三个SMN2副本的患者中保持运动功能。拷贝数对有症状患者治疗反应的影响尚不清楚。

ALS 和 SMA 的基因治疗
摘要:基因治疗是指通过施用遗传物质来修改、操纵基因表达或改变活细胞的特性以达到治疗目的。该领域的最新进展和改进已为各种疾病的治疗带来了许多突破。因此,人们对使用这些疗法治疗运动神经元疾病 (MND) 的兴趣日益浓厚,因为已发现了许多潜在的分子靶点。MND 是一种神经退行性疾病,在最严重的形式下,可导致呼吸衰竭和死亡,例如脊髓性肌萎缩症 (SMA) 或肌萎缩侧索硬化症 (ALS)。尽管 SMA 已为人所知多年,但它仍然是导致婴儿死亡的最常见遗传疾病之一。基于 ASO 的药物 nusinersen、小分子 risdiplam 和替代疗法 (GRT) — Zolgensma 的引入已在现有试验结果中显示出患者在使用这些疗法后无事件生存率和生活质量均有显着改善。尽管目前还没有药物能够有效缓解 ALS 的病程,但从 SMA 基因治疗中获得的经验为研制有效且安全的药物带来了希望。本综述旨在介绍基因治疗在 SMA 和 ALS 治疗中的当前进展和前景。

出显子7/外显子8缺失的患病率和脊柱肌肉萎缩症患者
脊柱肌肉萎缩(SMA)是由于脊髓前角细胞的变性而导致的神经肌肉疾病。SMA的估计发病率为1:6,000-1:10,000。SMN1基因的外显子7的完全缺失是大多数人群中95-98%的SMA患者的标志。对涉嫌患有SMA的儿童或年轻患者的第一条调查应为多重结扎依赖性探针扩增(MLPA)测试,用于对外显子7和SMN1基因中的外显子7和外显子8的纯合缺失。在本文中,我们报告了参加喀拉拉邦一家三级护理医院遗传诊所的SMN 1外显子7缺失测试的结果,其中有一个或多个症状,尤其是软盘婴儿,性低下,肌肉虚弱,舌头,舌头障碍等。SMN1外显子7和外显子8缺失在总计33例低调患者的58%(19)中得到了证实。在SMA阳性病例中,SMA I型,II型和III型分别为68.4%(13),21%(4)和10.5%(2)。对非语言父母的载体测试表明,所有父母都是杂合携带者。直到2016年,这种疾病的治疗仅是支持的。最近Nusinersen,Zolgensma和Risdiplam已适用于SMA患者。先前有SMA儿童历史的父母的携带者测试对于在未来怀孕中实施这种疾病的产前诊断至关重要。本文强调了这种罕见的神经肌肉疾病的重要性。

深入探索进展:脊髓性肌萎缩症当前治疗进展综述
脊髓性肌萎缩症 (SMA) 是一种罕见疾病,与基因有关,其特征是肌肉逐渐衰弱和退化,常常导致严重残疾和过早死亡。在过去十年中,SMA 治疗领域取得了显著进展,彻底改变了患者护理的格局。一项关键进展是基因靶向疗法的开发,例如 nusinersen、onasemnogene abeparvovec 和 risdiplam,它们在减缓疾病进展方面表现出前所未有的功效。这些疗法旨在通过靶向存活运动神经元 (SMN) 基因来解决 SMA 的根本原因,有效恢复缺陷的 SMN 蛋白水平。这些创新方法的出现改变了许多 SMA 患者的预后,为曾经治疗手段有限的患者带来了一线希望。此外,小分子化合物和 RNA 靶向策略的出现扩大了针对 SMA 的治疗手段。这些新干预措施表现出多种作用机制,包括 SMN 蛋白稳定和 RNA 剪接调节,展现了 SMA 治疗研究的多面性。制药行业、研究中心和患者权益团体的共同努力在加速将科学发现转化为可见的临床效益方面发挥了重要作用。这篇评论不仅突出了 SMA 治疗取得的显著进展,还为持续努力提高可及性、优化治疗策略、康复(护理和疗法)以及最终为改善 SMA 患者的生活质量铺平道路带来了希望之光。