XiaoMi-AI文件搜索系统
World File Search SystemP53
![技术数据表:293 [HEK-293] DCAS9-KRAB单元线](/simg/5\5845441ceb850691992d55c908fa0d66f7f3a759.webp)
技术数据表:293 [HEK-293] DCAS9-KRAB单元线
图2。在293 [HEK-293] DCAS9-KRAB细胞(ATCC®CRL-1573DCAS9-KRAB™)中对基因表达敲低的验证。抑制p53和setD9基因表达。表达靶向p53和setD9基因的GRNA的慢病毒分别用于感染293 [HEK-293] DCAS9-KRAB细胞。慢病毒没有GRNA表达作为对照。感染后24小时,将抗生素添加到培养基中,以富集抗生素耐药细胞。细胞颗粒并进行DDPCR基因表达定量分析。p53基因的表达(左)和setD9基因(右)在感染GRNA的细胞中受到了显着抑制。

MDM4异二聚体肽抑制剂可增强5 −
摘要:结直肠癌(CRC)通常涉及MDM2和MDM4过表达的野生型p53失活,从而促进了肿瘤的进展和对5-氟尿嘧啶(5-FU)的耐药性。破坏MDM2/4异二聚体可以熟练地重新激活p53,使癌细胞敏感到5-FU。在此,我们基于PEP3(1)开发了16种肽,这是唯一通过该机制作用的已知肽。新肽,尤其是3和9,与1相比,IC 50值较低。将纳米颗粒掺入肿瘤靶向的纳米颗粒中时,这些纳米颗粒对三种不同的CRC细胞系表现出细胞毒性。值得注意的是,NPS/ 9导致p53水平与其主要下游目标P21诱导细胞凋亡相关的p53水平显着增加。另外,9与5-FU的联合处理导致核仁应力的激活和协同凋亡效应。因此,MDM2/4异二聚体干扰物与5-FU通过纳米颗粒的共同传递可能是克服CRC中耐药性的有前途的策略。■简介

与肿瘤相关的抗原XCT和突变体-P53作为新组合抗肿瘤策略的分子靶标
摘要:胱氨酸/谷氨酸抗植物XCT是一种与肿瘤相关的抗原,在许多癌症类型中已被新近鉴定。通过参与谷胱甘肽生物合成,XCT可以保护癌细胞免受氧化应激条件和铁毒性的影响,并有助于代谢重编程,从而促进肿瘤的进展和化学抗性。此外,XCT在癌症干细胞中过表达。这些特征使XCT成为癌症治疗的有希望的靶标,正如文献中广泛报道的,在我们的免疫靶向方面。有趣的是,对TP53基因的研究表明,野生型和突变体p53均诱导了XCT的转录后下调调节,从而导致了铁毒性。APR-246是一种可以恢复癌细胞中野生型p53功能的小分子药物,已被描述为在具有突变体p53积累的肿瘤中XCT表达的间接调节剂,因此是一种与XCT抑制相结合的有希望的药物。本综述总结了当前对XCT的知识及其对p53的调节,重点是铁the虫中这两个分子的串扰,还考虑了一些可能的组合策略,这些策略可以与抗XCT免疫促进结合使用APR-246治疗。

CRISPR-Cas9 基因组编辑过程中致癌突变选择的系统性全基因组映射
sgRNA 而不是 NTC(图 3b,蓝线)。在涉及其他三个 CDE + 基因的竞争性测定中,未观察到 p53 WT 细胞中的反向富集(图 S6)。我们观察到,与 NTC 处理的细胞相比,靶向 NDUFB6 的 sgRNA 诱导的 DNA 损伤明显更高,特别是在 p53 WT 细胞中(图 S7),尽管突变细胞中的编辑效率更高(如图 4c 所示),这表明
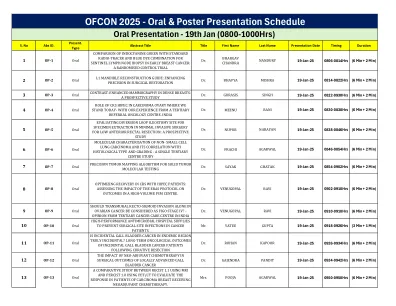
口头和海报演示时间表
3 pp-3 PIH1D1的海报作用R2TP复合物在p53和细胞周期调节中的稳定中。 Dhiraj Kumar Singh博士18-Jan-25 1300-1400hrs(3分钟 + 1分钟)3 pp-3 PIH1D1的海报作用R2TP复合物在p53和细胞周期调节中的稳定中。Dhiraj Kumar Singh博士18-Jan-25 1300-1400hrs(3分钟 + 1分钟)

MDM2 抑制联合内分泌治疗和 CDK4/6 抑制治疗 ER 阳性乳腺癌
背景:内分泌治疗耐药是雌激素受体 (ER) 阳性乳腺癌治疗的主要临床挑战。在这种情况下,p53 通常是野生型,其活性可能通过上调其关键调节因子 MDM2 而受到抑制。这构成了我们评估 MDM2 抑制作为治疗耐药性 ER 阳性乳腺癌的治疗策略的理论基础。方法:我们使用 MDM2 抑制剂 NVP-CGM097 单独治疗体外和体内模型以及与氟维司群或哌柏西利联合治疗。我们进行细胞活力、细胞周期、凋亡和衰老测定,以评估 p53 野生型和 p53 突变 ER 阳性细胞系 (MCF-7、ZR75-1、T-47D) 以及对内分泌治疗和 CDK4/6 抑制有耐药性的 MCF-7 系中的抗肿瘤作用。我们进一步评估了药物在内分泌敏感和内分泌抗性的 ER 阳性乳腺癌患者异种移植 (PDX) 模型中的作用。

针对 MDMX 进行癌症治疗:原理、策略......
致癌基因 MDMX(也称为 MDM4)是肿瘤抑制因子 p53 的关键负调节因子,与人类癌症的发生和发展有关。越来越多的证据表明,MDMX 通常在人类癌症中扩增并高度表达,促进癌细胞生长,并通过抑制 p53 介导的靶基因转录来抑制细胞凋亡。抑制 MDMX-p53 相互作用被发现可有效恢复 p53 的肿瘤抑制活性。因此,MDMX 正在成为开发抗癌疗法最有前途的分子靶点之一。在本综述中,我们主要关注当前的 MDMX 靶向策略和已知的 MDMX 抑制剂,以及它们的作用机制和体外和体内抗癌活性。我们还提出了其他潜在的靶向策略,以开发用于癌症治疗的更特异性和更有效的 MDMX 抑制剂。


针对去分化脂肪肉瘤中的 MDM2-p53 通路
去分化脂肪肉瘤 (DDLPS) 是一种恶性脂肪生成癌症,预后不良。DDLPS 肿瘤对化疗和放疗的敏感性较低,需要更有效的治疗方法。从遗传学上讲,DDLPS 的特点是肿瘤突变负担低,染色体结构异常频繁,包括 12q13-15 染色体区域和 MDM2 基因的扩增,这是 DDLPS 的定义特征。MDM2 蛋白是一种 E3 泛素连接酶,靶向肿瘤抑制因子 p53 进行蛋白酶体降解。人类恶性肿瘤中的 MDM2 扩增或过度表达与细胞周期进展和预后较差有关。因此,MDM2 - p53 相互作用作为 DDLPS 和其他恶性肿瘤的治疗靶点引起了人们的兴趣。MDM2 通过疏水蛋白相互作用与 p53 结合,这种相互作用很容易被合成类似物所利用。已经开发了多种药物,包括 Nutlins(如 RG7112)和小分子抑制剂(包括 SAR405838 和 HDM201)。临床前体外和动物模型已显示出抑制 MDM2 的良好效果,可导致 p53 强效再激活和癌细胞死亡。然而,多项早期临床试验未能证明抑制 MDM2 通路对 DDLPS 有益。耐药机制正在阐明,新型抑制剂和联合疗法目前正在研究中。本综述概述了针对 DDLPS 中 MDM2 的这些策略。

新型小分子 MDM2 抑制剂的设计和计算机模拟研究
摘要:蛋白质-蛋白质相互作用 (PPI) 在许多疾病状况中起着核心作用。因此,靶向和调节 PPI 是治疗疾病的有效方法。癌症也是由于蛋白质-蛋白质相互作用而产生的。在癌症中,肿瘤抑制因子 p53 蛋白被 MDM2 蛋白抑制。p53 蛋白调节细胞周期和细胞凋亡。p53-MDM2 蛋白之间的相互作用导致 p53 功能的抑制。通过这种相互作用,MDM2 降解并抑制 p53 蛋白。因此,靶向和抑制 p53-MDM2 相互作用来治疗癌症是合理的方法。通过用药物靶向这种相互作用,我们可以选择性地杀死癌细胞而不是正常细胞。最近,许多研究人员和制药公司已经报道了 p53-MDM2 相互作用抑制剂药物。并且有几种药物进入了临床试验。在这项研究中,一种新型的 1,2,4-三唑基分子被设计为 MDM2 抑制剂并进行了计算机研究。我们设计了新型化合物 01 和 Lead 1a。在这项工作中,对 Lead 1a 和参考化合物(Nutlin 3a、RG7112)进行了计算机模拟研究。对新型 1,2,4-三唑基 Lead 1a 和参考化合物进行了分子对接研究。发现 Lead 1a 的对接得分优于 Nutlin 3a 并且接近 RG7112。对接研究还确定了各种可能的构象和结合亲和力值。这些结果表明 Lead 1a 是一种潜在的 MDM2 抑制剂和抗癌剂。