XiaoMi-AI文件搜索系统
World File Search SystemPARP1
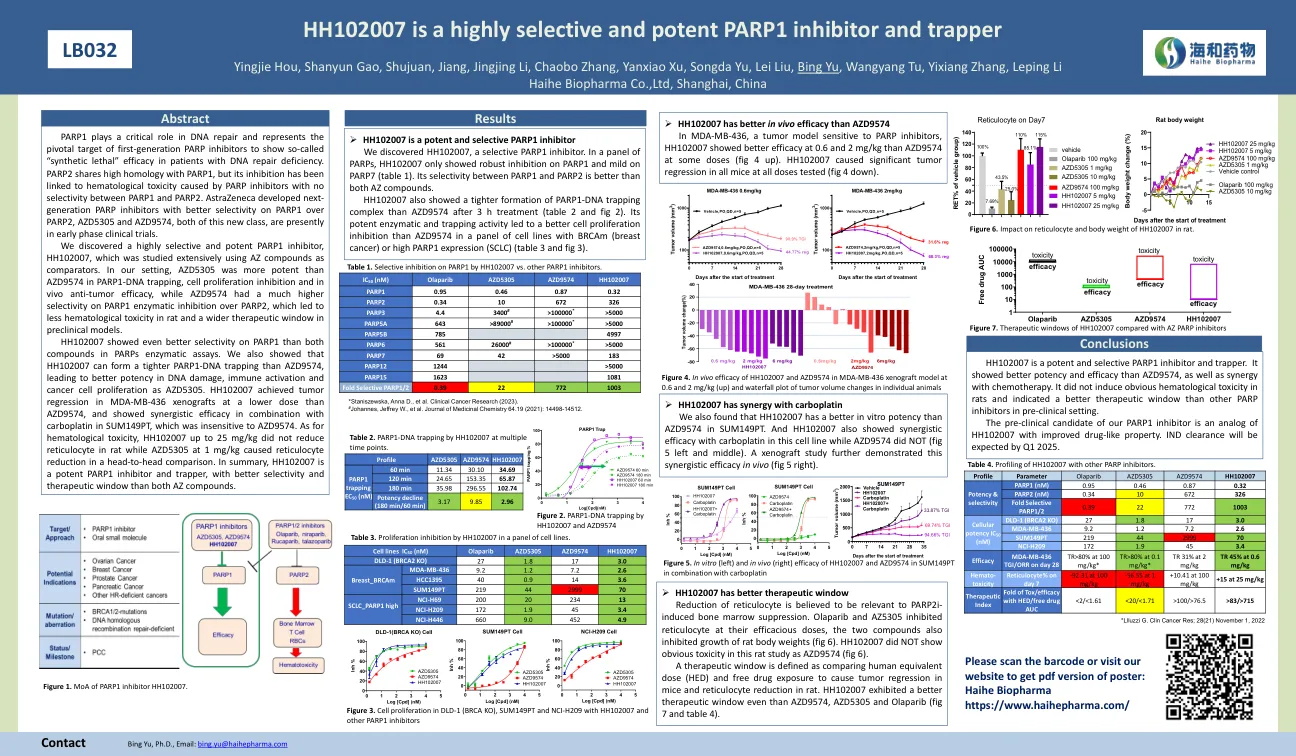
HH102007是一种高度选择性且有效的PARP1抑制剂和诱捕器
HH102007在PARP1上的选择性比PARPS酶测定中的两种化合物都更好。我们还表明,与AZD9574相比,HH102007可以形成更紧密的PARP1-DNA捕获,从而使DNA损伤,免疫激活和癌细胞增殖的效力更高,为AZD5305。HH102007在MDA-MB-436异种移植物中以低于AZD9574的剂量达到了肿瘤回归,并在SUM149PT中与卡波丁蛋白结合使用了协同功效,对AZD9574不敏感。至于血液学毒性,高达25 mg/kg的HH102007不会降低大鼠的网状细胞,而AZD5305的aZD5305在1 mg/kg时会导致头对头比较的网状细胞减少。总而言之,HH102007是一种有效的PARP1抑制剂和捕获器,比AZ化合物都具有更好的选择性和治疗窗口。

PARP1 调节乳腺癌中的 GATA3 介导的基因调控
系 生物医学科学 学位 哲学博士 在提交本论文以部分满足北达科他大学研究生学位的要求时,我同意本大学图书馆应免费提供本论文供查阅。我还同意,指导我论文的教授(如果教授不在,则由系主任或研究生院院长)允许我出于学术目的进行大量复制。双方了解,未经我的书面许可,不得为获取经济利益而复制、出版或以其他方式使用本论文或其中的任何部分。双方了解,在对我论文中的任何材料进行任何学术使用时,应给予我和北达科他大学应有的认可。

全面的机器学习增强了基于结构的PARP1抑制剂的虚拟筛查
聚ADP-核糖聚合酶1(PARP1)是癌症治疗的有吸引力的治疗靶标。机器学习评分功能构成了发现新型PARP1抑制剂的有前途方法。使用来自对接活性标记的分子的半合成训练数据研究了尖端PARP1特异性的机器学习评分功能:已知的PARP1抑制剂,与生成图神经网络并确认的Intactives consective contp1抑制剂,难以抗解的诱饵。我们仅包括与训练集中的分子不同,进一步使测试集更加困难。使用五种监督学习算法以及从对接姿势和配体中提取的蛋白质指纹对这些数据集的全面分析,只有两个高度预测性的评分功能。使用PARP1特异性支撑矢量的回归剂,使用PLEC指纹时,在最困难的测试集(NEF1%= 0.588,10个重复的中位数)中获得了高归一化富集因子,并且比其他任何研究的评分函数,尤其是类似的尺寸尺寸的尺寸。科学贡献

组蛋白 ADP 核糖基化促进 DNA 损伤中 PARP1 释放,从而增强对 PARP 抑制剂的耐药性
由于 PARP 抑制剂能够特异性地杀死无法通过同源重组修复 DNA 的肿瘤,因此聚(ADP - 核糖)聚合酶 1 (PARP1) 已成为癌症治疗的中心靶点。DNA 损伤后,PARP1 会迅速与 DNA 断裂结合并触发 ADP - 核糖基化信号。ADP - 核糖基化对于将各种因子募集到损伤部位以及及时将 PARP1 从 DNA 断裂中分离非常重要。事实上,在 PARP 抑制剂存在的情况下,PARP1 会被困在 DNA 断裂处,这是这些抑制剂细胞毒性的潜在机制。因此,任何影响捕获的细胞过程都被认为会影响 PARP 抑制剂的效率,可能导致接受这些药物治疗的患者产生获得性耐药性。DNA 损伤后有许多 ADP - 核糖基化靶点,包括 PARP1 本身以及组蛋白。虽然最近的研究报告称 PARP1 的自我修饰会促进其从 DNA 损伤中释放,但其他 ADP - 核糖基化蛋白对这一过程的潜在影响仍不清楚。本文,我们证明组蛋白 ADP - 核糖基化对于 PARP1 从损伤中及时消散也至关重要,从而导致细胞对 PARP 抑制剂产生耐药性。考虑到 ADP - 核糖基化与其他组蛋白标记之间的串扰,我们的研究结果为开发更有效的 PARP 抑制剂驱动的癌症疗法开辟了有趣的前景。

原创文章 AMXI-5001,一种用于治疗人类癌症的新型双重 parp1/2 和微管聚合抑制剂
摘要:聚(ADP-核糖)聚合酶 (PARP) 近来已成为癌症抵抗多种抗癌剂(包括微管靶向剂和 DNA 损伤剂等化疗剂)的中心介质。本文介绍了 AMXI-5001,这是一种新型、高效双重 PARP1/2 和微管聚合抑制剂,具有良好的代谢稳定性、口服生物利用度和药代动力学特性。通过生化分析确定了 AMXI-5001 的效力和选择性。体外评估了作为单一药物或与其他抗肿瘤药物联合使用的抗癌活性。在三阴性乳腺癌 (TNBC) 模型中评估了作为单一药物的体内抗肿瘤活性。AMXI-5001 对 PARP 和微管聚合的 IC50 抑制作用与临床 PARP 抑制剂(Olaparib、Rucaparib、Niraparib 和 Talazoparib)和强效聚合抑制剂(Vinblastine)相当。在体外,AMXI-5001 对多种人类癌细胞表现出选择性抗肿瘤细胞毒性,IC50 比现有的临床 PARP1/2 抑制剂低得多。AMXI-5001 在 BRCA 突变型和野生型癌症中均具有高度活性。AMXI-5001 可口服生物利用。AMXI-5001 在 BRCA 突变型 TNBC 模型中表现出显著的体内临床前抗肿瘤活性。口服 AMXI-5001 可诱导已建立的肿瘤完全消退,包括非常大的肿瘤。与单一药物(PARP 或微管)抑制剂或两种药物的组合相比,AMXI-5001 具有更优异的抗肿瘤效果。AMXI-5001 将很快进入临床试验测试,它代表了一种有前途的、新颖的同类首创的双重 PARP1/2 和微管聚合抑制剂,可通过一个分子提供连续和同步的一二连击癌症治疗。

2024; 20(5):1602-1616。 doi:10.7150/ijbs.85526研究论文PARP1促进心脏再生和心肌细胞增殖
心肌梗塞会导致心肌细胞丧失,并且出生后耗尽的心肌细胞增殖能力会影响心脏修复过程,最终导致心力衰竭。这项研究旨在研究聚(ADP-核糖)聚合酶1(PARP1)在心肌细胞增殖和心脏再生中的作用。我们的发现表明,PARP1敲除心肌细胞增殖,心脏功能和疤痕形成受损,而PARP1过表达改善了根尖切除术的小鼠的心脏再生。机械上,我们发现PARP1与热(ADP-核糖基)ates相互作用,热休克蛋白90 Alpha家族B成员1(HSP90AB1)与HSP90AB1和细胞分裂周期37(CDC37)(CDC37)和细胞周期酶活性之间的结合增加,因此激活了心脏模拟细胞细胞细胞周期。我们的结果表明,PARP1通过HSP90AB1的聚(ADP-核糖基)促进心脏再生和心肌细胞增殖,从而激活心肌细胞细胞周期,这表明PARP1可能是治疗心脏损伤的潜在治疗靶标。

肿瘤细胞中 MARCHF3 降解 PARP1 引发树突状细胞中 cCAS-STING 激活,从而调节肝细胞癌中的抗肿瘤免疫
摘要 背景 对免疫检查点抑制剂 (ICI) 的耐药性显著限制了肝细胞癌 (HCC) 患者免疫治疗的疗效。然而,免疫治疗耐药性的机制仍然不太清楚。我们的目的是在抗程序性细胞死亡蛋白 1 (PD-1) 治疗框架内阐明膜相关环 CH 型指 3 (MARCHF3) 在 HCC 中的作用。 方法 在对 ICI 表现出不同反应的 HCC 肿瘤的转录组谱中鉴定出 MARCHF3。在人类中,通过多重免疫组织化学评估 MARCHF3 表达与肿瘤微环境 (TME) 之间的相关性。此外,通过流式细胞术评估了肿瘤细胞中的 MARCHF3 表达和免疫细胞浸润。 结果 在对 ICI 有反应的患者的肿瘤中,MARCHF3 显著上调。HCC 细胞中 MARCHF3 表达的增加促进了树突状细胞 (DC) 成熟并刺激 CD8 + T 细胞活化,从而增强了肿瘤控制。从机制上看,我们确定 MARCHF3 是 DNA 损伤反应的关键调节因子。它通过 K48 连接的泛素化直接与聚(ADP-核糖)聚合酶 1 (PARP1) 相互作用,导致 PARP1 降解。该过程促进双链 DNA 的释放并激活 DC 中的 cCAS-STING,从而启动 DC 介导的抗原交叉呈递和 CD8 + T 细胞活化。此外,ATF4 转录调控 MARCHF3 表达。值得注意的是,PARP1 抑制剂奥拉帕尼增强了抗 PD-1 免疫疗法在皮下和原位 HCC 小鼠模型中的疗效。结论 MARCHF3 已成为 HCC TME 中免疫景观的关键调节因子,并且是 HCC 的有力预测生物标志物。将针对 DNA 损伤反应的干预措施与 ICI 相结合是一种有前途的 HCC 治疗策略。

doi:10.31557/apjcp.2025.26.2.611乳腺癌患者的PARP表达和糖酵解率限制酶的治疗靶向
背景:乳腺癌是一种异质性疾病,其特征是不同的生化,组织学和临床特征。PARP1和糖酵解速率限制酶在癌症进展中起关键作用,使它们成为有前途的治疗靶标。目的:本研究旨在评估乳腺癌患者中PARP1和关键糖酵解酶(HK,PFK和PK)的表达水平,并评估其作为治疗指标的潜力。材料和方法:研究中包括120名参与者(60名乳腺癌患者和60名健康对照组)。血液样本以测量使用ELISA的PARP1表达和糖酵解酶的水平。进行统计分析以比较两组。 结果:与健康对照组相比,乳腺癌患者的PARP1表达和糖酵解酶水平(HK,PFK和PK)明显更高(P <0.0001)。 结论:PARP1和关键糖酵解酶的过表达表明它们参与了乳腺癌的进展,并强调了它们作为治疗靶标和生物标志物的潜力。进行统计分析以比较两组。结果:与健康对照组相比,乳腺癌患者的PARP1表达和糖酵解酶水平(HK,PFK和PK)明显更高(P <0.0001)。结论:PARP1和关键糖酵解酶的过表达表明它们参与了乳腺癌的进展,并强调了它们作为治疗靶标和生物标志物的潜力。

组蛋白ADP-核糖化促进对PARP的抗性...
poly(ADP-核糖)聚合酶1(PARP1)由于PARP抑制剂特异性杀死通过同源重组而缺乏DNA修复的肿瘤的能力,因此已成为癌症疗法的核心靶标。在DNA损伤后,PARP1迅速与DNA断裂结合并触发ADP -Ribosylation信号传导。ADP-核糖基化对于募集各种因素到损害部位以及及时的DNA断裂中PARP1的分解很重要。的确,在存在PARP抑制剂的情况下,PARP1在DNA断裂处被困,这是这些抑制剂细胞毒素的基础机制。因此,任何影响捕获的细胞过程都被认为会影响PARP抑制剂效率,这可能会导致接受这些药物治疗的患者获得的耐药性。DNA损伤后有许多ADP-核糖基化靶标,包括PARP1本身以及组蛋白。最近的发现报道说,PARP1的自动修饰促进了其从DNA病变中释放,但其他ADP核糖基化蛋白对这一过程的潜在影响仍然未知。在这里,我们证明了组蛋白ADP - 核糖基化对于及时从病变中耗散PARP1的核糖基化也至关重要,从而有助于细胞对PARP抑制剂的耐药性。考虑ADP-核糖基化和其他组蛋白标记之间的串扰,我们的发现开辟了有趣的观点,可以开发出更有效的PARP抑制剂 - 驱动的癌症疗法。

PARP 抑制剂用于治疗小细胞肺癌及其与现有治疗方法整合的潜力
PARP 是一个蛋白质家族,它协调各种细胞过程,在 DNA 修复和基因组完整性方面发挥着重要作用。PARP1 可激活碱基切除修复 (BER),以响应 DNA 单链断裂 (SSB),其中 PARP1 与 SSB 结合并促进 DNA 修复蛋白的募集。当 PARP1 功能受损时,BER 过程会停止,并且由于复制叉不稳定而导致双链断裂 (DSB) 发生 (18)。因此,缺乏同源重组 (HR) DSB 修复途径的恶性肿瘤容易受到 PARP 抑制。PARPi 首次被证明对 BRCA1/2 突变的卵巢癌有效,而这些卵巢癌缺乏 HR (19)。随后,PARPi 的临床疗效扩展到其他携带 BRCA1/2 突变的组织学(19-27),其中大多数 PARPi 获得 FDA 批准用于治疗 BRCA1/2 突变的卵巢癌和乳腺癌(表 1)(30-37)。