XiaoMi-AI文件搜索系统
World File Search SystemPARP1
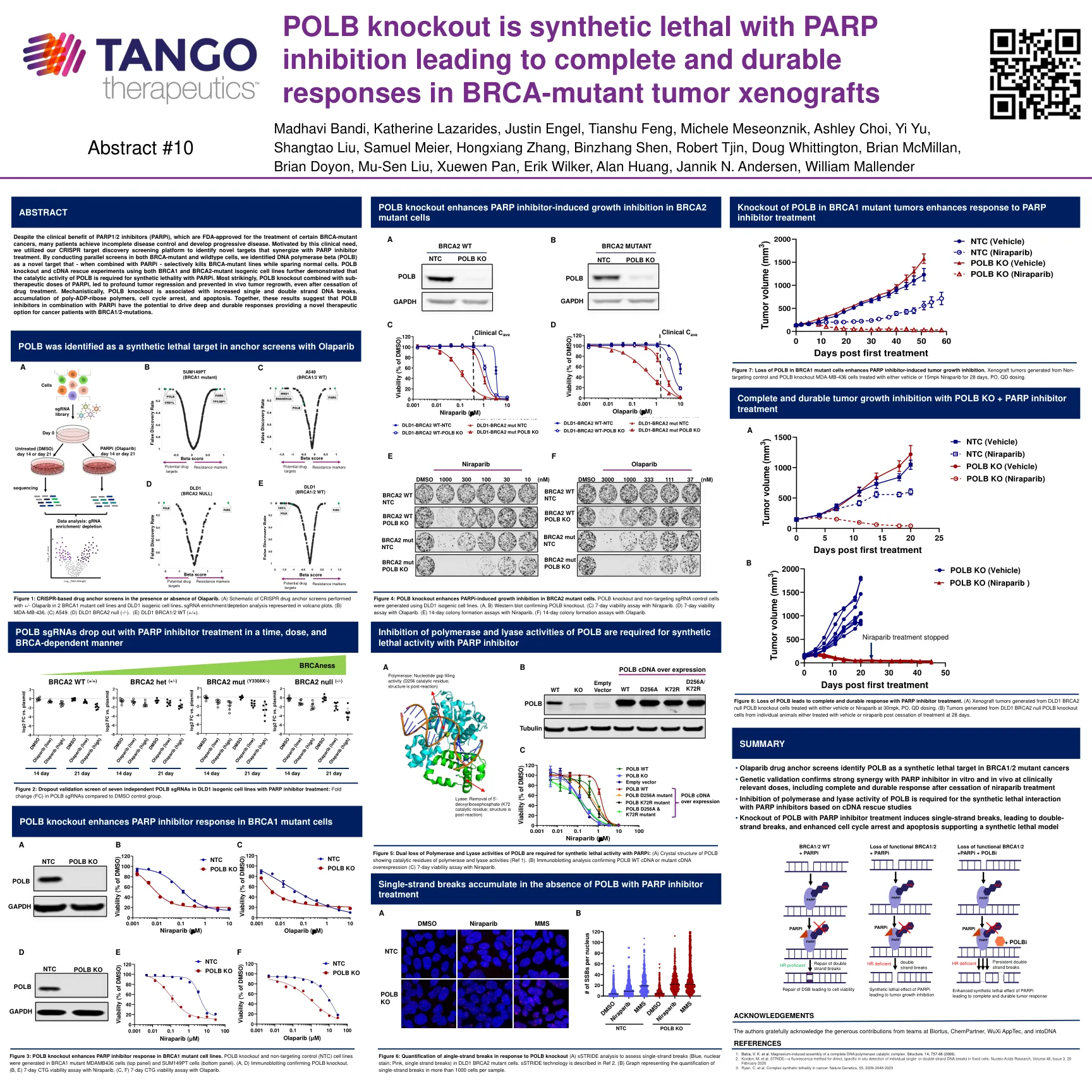
POLB敲除具有合成的致死性,并抑制了PARP,从而导致BRCA-突变肿瘤异种移植物的完整耐用反应
尽管PARP1/2抑制剂(PARPI)的临床益处是FDA批准用于治疗某些BRCA-突变癌的临床益处,但许多患者可以实现不完全的疾病控制和发展性疾病。是出于这种临床需求的激励,我们利用了CRISPR目标发现筛选平台来确定与PARP抑制剂治疗协同作用的新目标。通过在BRCA-突变剂和野生型细胞中进行平行筛选,我们将DNA聚合酶β(POLB)鉴定为一个新靶标,当与PARPI结合使用时,可以选择地杀死BRCA突变线,同时放大正常细胞。POLB敲除和使用BRCA1和BRCA2突变的同基因细胞系的cDNA救援实验进一步证明,PORB的催化活性对于与PARPI合成的致死性是必需的。最引人注目的是,POLB敲除与亚治疗剂量的PARPI结合,导致了深层肿瘤的消退,并阻止了体内肿瘤再生,即使停止药物治疗。从机械上讲,polb敲除与单链DNA断裂增加,多-ADP-核糖聚合物的积累,细胞周期停滞和凋亡有关。在一起,这些结果表明,POLB抑制剂与PARPI结合使用,有可能推动深层耐用的反应,为BRCA1/2突变的癌症患者提供了一种新型的治疗选择。

lig1失活选择性地抑制BRCA1突变细胞在体外和体内的生长
我们在11个BRCA1/2野生型和2个BRCA1突变癌细胞系中进行了CRISPR筛选,以识别与BRCA1突变合成致死的靶标。除了USP1,PARP1和POLQ外,我们还将DNA连接酶基因lig1确定为新的靶标,当被击倒后,会选择性地杀死BRCA1突变细胞。对乳腺癌和卵巢癌细胞系中Achilles数据库中BRCA突变的内部分析进一步验证了BRCA1突变细胞在LIG1上的超依赖性。使用CRISPRN,CRISPRI和RNAI对LIG1的单基因扰动证实了LIG1在BRCA1突变细胞系中失活的致命作用,但不能BRCA1/2野生型细胞系。可以通过与外源性野生型LIG1 cDNA相辅相成,证明遗传工具的目标性质可以挽救这种生存能力。使用可降解的DNA连接酶I融合蛋白,我们证明了DNA连接酶I蛋白水平与BRCA1突变细胞中的生存能力之间存在很强的相关性。酶上无活性的DNA连接酶I突变蛋白(LIG1 K568A)无法营救由内源性LIG1耗竭引起的生存力的损失,从而从小分子抑制剂的角度来支持该靶标的易生化性。使用BRCA1突变体MDA-MB-436衍生的肿瘤在体内复制这些数据,其中肿瘤生长在LIG1丢失后抑制了> 80%。

DNA 损伤反应抑制剂增强胶质瘤干细胞中肿瘤治疗场 (TTFields) 的效力
背景:高级别胶质瘤是原发性脑癌,过去 40 年来,尽管进行了手术切除和破坏 DNA 的化放疗,但世界卫生组织 4 级胶质瘤的生存率仍然低得令人无法接受,且持续为 10-16 个月。最近,肿瘤治疗场疗法 (TTFields) 已显示出适度的生存益处,并在多个国家获得临床批准。TTFields 人们认为主要通过破坏有丝分裂来介导抗癌活性。然而,最近的数据表明,TTFields 也可能减弱 DNA 损伤修复和复制叉动力学,为结合标准治疗方法和靶向 DNA 损伤反应抑制剂 (DDRi) 的治疗组合提供了潜在的平台。方法:我们将患者来源的、通常具有抗性的胶质瘤干细胞样细胞 (GSC) 与之前验证的临床前 Inovitro™ TTFields 系统以及多种治疗性 DDRi 结合使用。结果:我们发现 TTFields 可强效激活 PARP 和 ATR 介导的 DNA 修复(包括 PARylation 和 CHK1 磷酸化),而将 TTFields 与 PARP1 或 ATR 抑制剂治疗相结合可显著降低克隆形成存活率。放射治疗进一步增强了这些策略的效力,导致 DNA 损伤量增加,DNA 损伤消退时间大大延迟。结论:据我们所知,我们的研究结果是首次在 GSC 模型中将 TTFields 与临床批准或试验中的 DDRi 结合使用,并为针对目前无法治愈的肿瘤患者的多模式 DDRi/TTFields 治疗策略的转化研究提供了基础。

研究论文 lncRNA MNX1-AS1下调促进非小细胞肺癌铁死亡和细胞凋亡
非小细胞肺癌(NSCLC)是肺癌的主要组织学类型,对人类健康构成严重威胁。越来越多的证据表明,长链非编码RNA(lncRNA)MNX1-AS1参与了癌症(包括肺癌)的发生发展。细胞凋亡和铁死亡是两种受调控的细胞死亡形式,可由抗癌药物诱导。然而,MNX1-AS1在细胞凋亡和铁死亡中的作用尚不清楚。本文我们发现,敲低MNX1-AS1可促进RSL3诱导的NSCLC细胞铁死亡,导致细胞活力下降,活性氧(ROS)和丙二醛(MDA)水平升高。吖啶橙/溴化乙锭(AO/EB)双染、末端脱氧核苷酸转移酶介导的dUTP缺口末端标记(TUNEL)实验及Annexin V/PI双染实验均显示敲低MNX1-AS1可促进紫杉醇诱导的NSCLC细胞凋亡。此外,敲低MNX1-AS1还导致促凋亡蛋白BAX、cleaved caspase-3及PARP1表达增加,抗凋亡蛋白Bcl-2表达减少。RNA测序及实时荧光定量PCR检测发现,敲低MNX1-AS1后,ACSL4表达增加,而ABCG2表达减少。挽救实验显示,ACSL4和ABCG2分别参与了MNX1-AS1介导的铁死亡和细胞凋亡。此外,敲低 MNX1-AS1 可增加 NSCLC 细胞对 RSL3 和紫杉醇组合的敏感性。总之,我们的数据表明 MNX1-AS1 可能是肺癌的潜在治疗靶点,尤其是与铁死亡和/或凋亡诱导药物组合使用时。

BH3 模拟药物与替莫唑胺、JQ1 和铁死亡诱导剂协同杀死多形性胶质母细胞瘤细胞
多形性胶质母细胞瘤 (GBM) 是最常见且最具侵袭性的脑癌,由于恶性细胞对常规疗法具有固有的耐药性,治疗选择通常受到限制。我们研究了使用 BH3 模拟药物在人类 GBM 细胞系中触发程序性细胞死亡 (PCD) 的影响。我们证明,与使用替莫唑胺或溴结构域抑制剂 JQ1 的常规体外疗法相比,同时靶向促存活蛋白 BCL-XL 和 MCL-1 可更有效地杀死六种 GBM 细胞系。与单一药物治疗相比,在使用 TMZ 或 JQ1 联合 BCL-XL 抑制剂的双重治疗下,U251 和 SNB-19 细胞中观察到细胞杀伤力增强。这反映在 caspase-3 的大量裂解/活化以及 PARP1 的裂解(凋亡标志物)中。与使用 BCL-2 抑制剂 Venetoclax 和 BCL-XL 抑制剂的双重治疗相比,使用针对 BCL-XL 和 MCL-1 的 BH3 模拟物组合更容易杀死 U251 和 SNB-19 细胞。BAX 和 BAK(内在凋亡的基本执行者)的共同丧失使 U251 和 SNB-19 细胞对任何测试的药物组合都具有抗药性,表明凋亡是导致它们死亡的原因。在 GBM 的原位小鼠模型中,我们证明 BCL-XL 抑制剂 A1331852 可以渗透到大脑中,在肿瘤和健康大脑区域均检测到 A1331852。我们还研究了将铁死亡的小分子诱导剂 erastin 和 RSL3 与 BH3 模拟药物相结合的影响。我们发现 BCL-XL 或 MCL-1 抑制剂可与铁死亡诱导剂有效协同杀死 U251 细胞。总体而言,这些发现证明了双重靶向 GBM 中不同 PCD 信号通路的潜力,并可能指导 BCL-XL 抑制剂和铁死亡诱导剂与标准护理治疗的结合使用,以改善 GBM 疗法。

蕨二醇通过激活 MAPK/ERK 通路诱导肝细胞癌细胞凋亡
肝细胞癌(HCC)具有较高的致死率和致残率,严重危害人类的生命。化学药物和化疗药物在HCC治疗中一直存在副作用、耐药性等问题,不能满足临床治疗的需要。因此寻找新型低毒高效的抗肝细胞癌药物并探究其作用机制成为当前HCC治疗中亟待解决的问题。已有多项研究报道了inotodiol的抗癌作用,本研究针对inotodiol在HCC细胞中的抗癌作用及其分子机制,旨在深入探究其抗癌作用。采用CCK8实验检测细胞存活率,划痕实验检测细胞迁移能力,克隆形成实验检测克隆形成能力,流式细胞术分析细胞凋亡和细胞周期。通过动物实验验证inotodiol对HCC的抑制作用。同时采用western blotting检测凋亡、细胞周期及MAPK/ERK通路相关蛋白。结果表明inotodiol具有促进细胞凋亡、抑制细胞增殖、迁移和克隆形成的能力,当CDK2、CDK4、CDK6和Cyclin D的表达受到抑制时,细胞周期被阻滞在G1期。此外,inotodiol表现出诱导细胞凋亡的作用,其特点是Bax表达增加,Bcl-2、Bcl-XL和MCL1表达减少,PARP1和caspase 3的剪切启动,以及MAPK/ERK通路的抑制。动物实验表明inotodiol具有抑制裸鼠肿瘤生长的能力,同时对小鼠的体重和脏器无明显影响。总之,本文提出的研究结果有力地表明,inotodiol 可以成为治疗肝细胞癌 (HCC) 的有希望的候选药物。

PARP抑制剂和蛋白质与SLX4相互作用
PARP-1蛋白通过将XRCC1募集到修饰的DNA位置来参与单链断裂修复。当抑制PARP时,细胞依赖其他DNA修复机制,尤其是同源重组,以正确复制基因组信息,而无需进行致命性有丝线的风险。在具有同源重新组合的细胞中,例如BRCA1-或BRCA2突变的细胞,PARP抑制是致命的[1,2]。在2005年提供了这些描述后,合成致死性的概念出现,而PARP抑制剂(PARPI)的开发是为了治疗BRCA-Muthated患者,在该患者中,非癌细胞具有一个野生型等位基因,而癌细胞则是BRCA的定义,因此是特异性敏感的,因此具有特异性敏感性。几个PARPI已在临床上进行了研究,可用于治疗癌症患者(Olaparib,Rucaparib,Talazoparib,Niraparib和Veliparib(ABT-888))。有关PARPI的科学文献非常丰富(自2005年以来> 12,000篇论文),研究论文,临床试验和评论涉及有关作用机理,抗药性,临床活动以及新化合物的发展。最初认为PARPI的作用机理是对PARP1相关的单链破裂修复的“简单”抑制作用,随后出现更具毒性和更容易恢复的双链断裂。然而,真理要复杂得多,正如T. Helleday [3]已经讨论的那样,自从该出版物[4]开始。关于Parpi的许多知识仍然未知,它们的临床可能比今天所描述的要强。基于这些知识的工作促进了与PARPI活性和耐药机制有关的其他蛋白质的鉴定,并有助于发展与其他DNA相关蛋白(如RAD51 [5]和EZH2 [6]的药理抑制其他与DNA相关蛋白的相关策略[6]。特别是其他DNA修复的可能参与
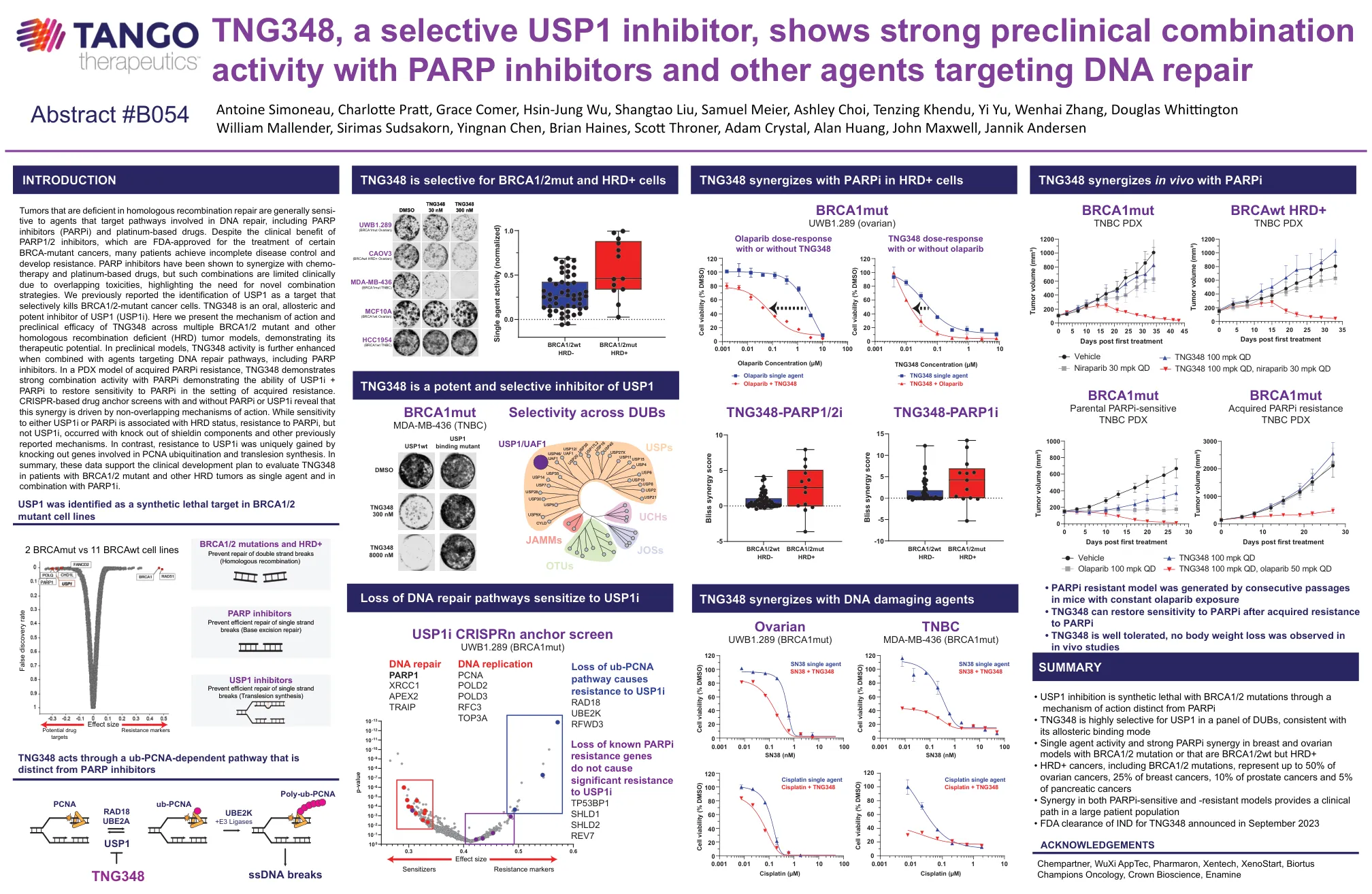
TNG348 海报 ENA 2023Final
同源重组修复缺陷的肿瘤通常对针对 DNA 修复途径的药物敏感,包括 PARP 抑制剂 (PARPi) 和铂类药物。尽管 PARP1/2 抑制剂具有临床益处,且已获 FDA 批准用于治疗某些 BRCA 突变癌症,但许多患者的疾病控制并不完全并且产生了耐药性。PARP 抑制剂已被证明可与化疗和铂类药物产生协同作用,但由于毒性重叠,此类组合在临床上受到限制,这凸显了对新组合策略的需求。我们之前报告已鉴定出 USP1 可选择性杀死 BRCA1/2 突变癌细胞的靶点。TNG348 是一种口服、变构和强效的 USP1 (USP1i) 抑制剂。这里我们介绍了 TNG348 在多种 BRCA1/2 突变体和其他同源重组缺陷 (HRD) 肿瘤模型中的作用机制和临床前疗效,证明了其治疗潜力。在临床前模型中,当与针对 DNA 修复途径的药物(包括 PARP 抑制剂)结合时,TNG348 活性会进一步增强。在获得性 PARPi 耐药性的 PDX 模型中,TNG348 表现出与 PARPi 的强大组合活性,证明了 USP1i + PARPi 能够在获得性耐药的情况下恢复对 PARPi 的敏感性。有和没有 PARPi 或 USP1i 的基于 CRISPR 的药物锚定筛选表明,这种协同作用是由不重叠的作用机制驱动的。虽然对 USP1i 或 PARPi 的敏感性与 HRD 状态有关,但对 PARPi 而非 USP1i 的耐药性发生在屏蔽蛋白成分的敲除和其他先前报道的机制下。相比之下,通过敲除参与 PCNA 泛素化和跨损伤合成的基因,可以独特地获得对 USP1i 的抗性。总之,这些数据支持临床开发计划,以评估 TNG348 作为单一药物和与 PARP1i 联合用于 BRCA1/2 突变体和其他 HRD 肿瘤患者。

12:30 PM-4:00 PM 2级,展览馆D A001:MAPP
海报会议C 10月14日,星期六| 12:30 pm-4:00 PM 2级,展览馆D LB_C02:AV-380与Cachexia的转移性癌症患者(PTS)结合使用AV-380的1B期剂量升级研究和GDF-15升高。马丁·伯克霍夫(Martin Birkhofer),美国马萨诸塞州波士顿Aveo肿瘤学。LB_C03:蛋白质翻译抑制作用会强制组蛋白脱乙酰基酶抑制剂活性,从而导致协同胰腺癌细胞死亡。Maryam Safari,美国纽约哥伦比亚大学医学中心。LB_C04:新型的口服生物利用的大环,靶向细胞周期蛋白A和B在乳腺癌患者衍生的异种移植模型中引起抗肿瘤活性。Mariana Paes Dias,Vall D'Hebron肿瘤学研究所,西班牙巴塞罗那。lb_c05:一种新的方法,是通过血管靶向的光动力疗法对帕德氏菌素内血管内激活进行主要动脉参与的不可切除的胰腺癌的新方法。dina Preise,Impact Biotech Ltd,韦兹曼科学学院,内斯·西奥纳(Ness Siona),以色列Rehovot。LB_C06:利用新型的HDAC抑制剂Bocodepsin(OKI-179)克服三阴性乳腺癌中的阿霉素耐药性。Stephen G. Smoots,Cu Anschutz,美国丹佛,美国。lb_c07:利用Bcl-2抑制剂(Venetoclax)克服三阴性乳腺癌中的阿霉素耐药性。埃文·杜斯(Evan Dus),科罗拉多州科罗拉多大学,美国阿罗拉,美国。LB_C09:QTX3034,一种有效的多KRAS抑制剂,与EGFR抑制剂协同作用,并增强了抗肿瘤活性。Sarah Truong,Rakovina Therapeutics,不列颠哥伦比亚省加拿大温哥华。Jillian M. Silva,Quanta Therapeutics,南旧金山,美国加利福尼亚州。 lb_c10:一种口服的小分子抑制剂,用于合成MYC表达肿瘤的致命靶向。 Thaddeus D. Allen,抗癌Bioscience,Inc。,美国加利福尼亚州旧金山。 lb_c11:PARP1/2和HDAC酶的小分子双功能抑制剂。 lb_c12:Alisertib和pembrolizumab在RB缺陷的头部和颈部鳞状细胞癌(HNSCC)中。 Faye M. Johnson,德克萨斯大学医学博士安德森癌症中心,美国德克萨斯州休斯敦。 LB_C13:BLX-3030的开发,一种有效的,有选择性的口服CDK9I在胰腺导管腺癌(PDAC)模型中显示出希望。 凯尔·梅德利(Kyle Medley),美国美国叉子(American Fork),美国叉子(American Fork),美国。 LB_C14:EAI-432:一种用于L858R突变的非小细胞肺癌的突变选择性变构EGFR抑制剂。 迈克尔·J·埃克(Michael J.Jillian M. Silva,Quanta Therapeutics,南旧金山,美国加利福尼亚州。lb_c10:一种口服的小分子抑制剂,用于合成MYC表达肿瘤的致命靶向。Thaddeus D. Allen,抗癌Bioscience,Inc。,美国加利福尼亚州旧金山。lb_c11:PARP1/2和HDAC酶的小分子双功能抑制剂。lb_c12:Alisertib和pembrolizumab在RB缺陷的头部和颈部鳞状细胞癌(HNSCC)中。Faye M. Johnson,德克萨斯大学医学博士安德森癌症中心,美国德克萨斯州休斯敦。 LB_C13:BLX-3030的开发,一种有效的,有选择性的口服CDK9I在胰腺导管腺癌(PDAC)模型中显示出希望。 凯尔·梅德利(Kyle Medley),美国美国叉子(American Fork),美国叉子(American Fork),美国。 LB_C14:EAI-432:一种用于L858R突变的非小细胞肺癌的突变选择性变构EGFR抑制剂。 迈克尔·J·埃克(Michael J.Faye M. Johnson,德克萨斯大学医学博士安德森癌症中心,美国德克萨斯州休斯敦。LB_C13:BLX-3030的开发,一种有效的,有选择性的口服CDK9I在胰腺导管腺癌(PDAC)模型中显示出希望。凯尔·梅德利(Kyle Medley),美国美国叉子(American Fork),美国叉子(American Fork),美国。LB_C14:EAI-432:一种用于L858R突变的非小细胞肺癌的突变选择性变构EGFR抑制剂。迈克尔·J·埃克(Michael J.

DNA损伤,修复和癌症代谢
DNA损伤反应(DDR)与代谢之间的复杂相互作用,对管理基因组完整性维持的基本机制有深刻的了解[1]。细胞不断遇到诱导DNA损伤的内源性和外源性威胁,如果未修复,可能会导致突变,基因组不稳定性,并最终导致癌症等疾病[2]。代谢为DNA修复过程提供了必要的能量和构建块[3]。值得注意的是,DDR和代谢中的关键信号通路和酶促活性都紧密相关。例如,ATM和ATR激酶对DNA损伤的激活直接通过调节MTOR途径和细胞能量来直接影响细胞代谢状态[4]。此外,DNA修复酶(例如PARP1)与NAD+代谢相关,其活性会影响细胞生物能学[5]。DDR和代谢之间的这种复杂的串扰不仅确保基因组稳定性,而且还低估了细胞稳态在保护遗传信息中的重要作用,这使其成为对人类健康和疾病有深远影响的关键研究领域。本期特刊介绍了DNA损伤反应和癌症代谢领域领先专家的九篇论文。这些论文重点介绍了特定DNA破坏药物的药代动力学和药效学分析的最新进展,以及在DDR中发现新因素和调节机制的发现,包括DNA修复,检查点途径,复制应激,细胞死亡,细胞死亡和癌症代谢。Park等。Park等。此外,这些论文阐明了这些系统之间复杂的串扰,为基因组稳定性和针对DNA损伤的细胞代谢的广泛景观提供了宝贵的见解。在依托泊苷(ETO)处理中探究锌纤维蛋白Zatt的作用,揭示其在修复拓扑异构酶II(TOP2)的双重功能 - DNA共价复合物(TOP2CC)并在ETO治疗后促进细胞存活。ETO稳定瞬态top2cc,导致DNA双链断裂(DSB)。TOP2CC的修复涉及酪酶-DNA磷酸二酯酶2(TDP2),它从DSB的5'末端去除磷酸酪糖基肽。这项研究采用了全基因组CRISPR筛选,并证明Zatt在ETO处理后促进细胞存活中起着至关重要的作用,与TDP2-KO细胞相比,Zatt敲除(KO)细胞显示对ETO的敏感性提高。对Zatt的结构方面的进一步研究表明,N末端1-168残基对于与TOP2相互作用至关重要,显着影响ETO敏感性。在ETO或环己二酰亚胺处理后加速了TOP2降解,表明其在提高TOP2稳定性的作用,并可能导致TOP2周转率。这些发现表明,Zatt是对ETO治疗的反应的关键决定因素,其承诺是增强ETO在癌症治疗中效率的策略。Yeom等。 研究了与DNA聚合酶η相关的三种人Polh种系变体的功能特性,DNA聚合酶η是一种关键酶,负责无错误的跨性别DNA合成(TLS)。Yeom等。研究了与DNA聚合酶η相关的三种人Polh种系变体的功能特性,DNA聚合酶η是一种关键酶,负责无错误的跨性别DNA合成(TLS)。这些变体与易皮肤癌的结合(即,静脉表色素变体(XPV))和对顺铂的敏感性增加。生化和基于细胞的测定法用于评估这些种系的影响