XiaoMi-AI文件搜索系统
World File Search SystemProtein

蛋白质合成流图
蛋白质合成是在所有生物体中发生的重要细胞过程,涉及蛋白质的产生。此复杂的过程由两个阶段组成:转录和翻译。转录发生在细胞核内,DNA充当产生信使RNA的模板(mRNA)。mRNA然后传播到细胞质的核糖体,这是翻译的位置。在这里,mRNA携带的遗传信息被解码以合成多肽链。**转录**是蛋白质合成的初始阶段,其中DNA的遗传密码被转录为mRNA。当RNA聚合酶附着在基因的启动子序列上时,此过程就开始了,促使DNA放松。酶然后读取DNA碱基并组装互补的mRNA链。用作模板的DNA链被称为模板或反义链,而其对应物是非编码或感官链。新形成的mRNA链反射了编码DNA链,尿嘧啶代替了胸腺素。**处理mRNA **涉及新合成的mRNA的进一步细化,也称为前mRNA。在它可以将细胞核作为成熟的mRNA退出之前,它会经历剪接,编辑和聚腺苷酸化,从而改变mRNA以准备翻译。对于有兴趣可视化此过程的人,**蛋白质合成流程图**可以是一个有用的工具。它提供了从DNA转录到最终蛋白质产物的蛋白质合成每个步骤的清晰结构化表示。此外,mRNA经过编辑,改变了某些核苷酸。这样的流程图可以帮助理解基于这种基本生物学功能的复杂相互作用和机制。遗传修饰增强了单个基因的多功能性,使其能够产生多种蛋白质。这是通过称为剪接的过程来实现的,该过程从蛋白质合成流程图中描述了从信使RNA(mRNA)中去除被称为内含子的非编码区域。剪接的mRNA仅由编码区域或外显子组成,这直接有助于蛋白质合成。核糖核蛋白,核中含有RNA的小蛋白,可促进该剪接。例如,由于这种编辑,参与血液中脂质转运的APOB蛋白以两种形式存在。较小的变体是由于插入的停止信号截断了mRNA的插入信号。5'上限过程为mRNA的铅端增加了一个保护性的甲基化盖,从而保护了它免于降解和辅助核糖体附着。一系列腺嘌呤碱基的尾巴标志着mRNA的结论,在其核出口和防御降解酶的防御中发挥了作用。分子生物学的中心教条概述了从RNA到蛋白质的过渡,这一过程称为翻译。这涉及将mRNA中的遗传代码读取以合成蛋白质,如流程图所示。后加工,mRNA将核和核糖体缔合,由核糖体RNA(rRNA)和蛋白质组成。核糖体解密mRNA序列,而转移RNA(tRNA)分子依次传递适当的氨基酸。翻译分为三个阶段:启动,伸长和终止。在开始期间,现在在细胞质中的mRNA与甲基化帽和起始密码子位点的核糖体亚基结合。具有与起始密码子连接的具有匹配的反物质的tRNA,形成了起始复合物。伸长涉及连续供应氨基酸的TRNA,这些氨基酸被添加到新生的多肽链中。每个tRNA转移后其氨基酸后出发,使核糖体沿mRNA进行进展,从而为下一个tRNA腾出空间。这种系统的添加氨基酸构建了多肽,直到该过程结束为止。蛋白质合成是一个重要的细胞过程,最终导致蛋白质的产生。它在两个主要阶段展开:转录和翻译。在转录过程中,DNA的遗传密码被转录为核中的信使RNA(mRNA),包括三个阶段:启动,伸长和终止。mRNA然后将这些遗传指令传输到发生翻译的细胞质核糖体。由核糖体RNA(RRNA)和蛋白质组成的核糖体读取mRNA序列。转移RNA(tRNA)分子根据mRNA代码将适当的氨基酸带入核糖体。rRNA促进了这些氨基酸的粘结,形成了多肽链。该链可能会进一步进行合成后修饰以实现其最终蛋白质结构。mRNA退出核之前,它会经过加工,成为准备翻译的成熟转录本。蛋白质合成的过程与分子生物学的中心教条一致,该过程映射了生物系统中遗传信息的流动。合成后,多肽链可能会折叠成特定的形状,与其他分子相互作用,或在内质网中进行其他修饰以实现其指定的功能。

工业生物技术中的蛋白质工程
本书分别分为三个部分:用于生物催化和医疗保健的蛋白质工程。涵盖了更相关的工业酶:脂肪酶,蛋白酶,羧肽酶,葡萄糖酶和葡萄糖酶,果胶分解酶和酶,用于重新固定化合物的生物化。还讨论了溶剂工程的相互作用以调节结构 - 活动关系。一章致力于将蛋白质工程应用于生物传感器。

肿瘤靶向蛋白的延长释放——...
1 巴塞罗那自治大学生物技术与生物医学研究所,Cerdanyola del Vallès,08193 巴塞罗那,西班牙; srnaroa@gmail.com(国家安全); carlosmartineztorro@gmail.com(CM-T.); Esther.Vazquez@uab.cat (EV) 2 巴塞罗那自治大学遗传学和微生物学系,Cerdanyola del Vallès,08193 巴塞罗那,西班牙; uunzueta@santpau.cat 3 CIBER de Bioingenieri í a, Biomateriales y Nanomedicina (CIBER-BBN), 28029 马德里, 西班牙; AFalgas@santpau.cat (AF); agarciale@santpau.cat(AG-L.); YNunez@santpau.cat (YN); rmangues@santpau.cat (RM) 4 圣保罗生物医学研究所(IIB Sant Pau),圣克鲁伊圣保罗医院,08025 巴塞罗那,西班牙 5 Josep Carreras 研究所,巴达洛纳,08916 巴塞罗那,西班牙 6 Servei de Pia,贝拉大学,巴塞罗那 08193 巴塞罗那,西班牙; Alejandro.Sanchez.Chardi@uab.cat 7 巴塞罗那大学生物学院进化生物学、生态学和环境科学系,Av. Diagonal 643, 08028 巴塞罗那,西班牙 * 通讯地址:ICasanova@santpau.cat (IC); Antoni.villaverde@uab.cat (AV) † 这些作者对这项工作做出了同等贡献。 ‡ 现地址:Nanoligent SL。西班牙巴塞罗那自治大学尤里卡大楼,贝拉特拉,08193 巴塞罗那,邮编。

用于蛋白质和肽递送的纳米载体
摘要:蛋白质和多肽已被公认为合成治疗多种人类疾病的新型疗法的潜在线索。不幸的是,由于递送应用的边缘性,这些生物大分子的治疗潜力和临床应用具有挑战性。纳米载体具有独特的潜力,可以克服各种生物屏障并改善蛋白质和多肽等治疗性生物大分子的递送。基于智能纳米载体的药物递送系统可以定义为一种以受控方式针对所有生理屏障进行位点特异性药物递送并最终在体内代谢的系统。本综述介绍了用于递送蛋白质和多肽以增强其临床应用的各种纳米载体。我们还重点介绍了蛋白质和多肽递送的各种生物学方面。我们还总结了用于递送这些生物大分子的纳米载体的各种专利,然后概述了用于递送蛋白质和多肽的市售纳米制剂。

马尼托巴蛋白质研究战略
图 3 显示了调查受访者的研究领域。53% 的调查受访者正在开展初级生产/收获方面的研究,其次是 35% 的受访者在加工和食品安全或营养评估方面进行研究。共有 54 名研究人员(占调查参与者的 47%)正在蛋白质食品系统的多个领域进行研究。一些研究人员表示,他们在其他研究领域进行研究,包括环境可持续性、土地和水管理、数字农业、作物遗传学、土壤科学、社会经济学、粮食主权、风味组学、生物材料和传染病。

了解目标蛋白质降解
1. 什么是蛋白质?它们有什么作用?:MedlinePlus Genetics。访问日期:2021 年 5 月 3 日。https://medlineplus.gov/genetics/understanding/howgeneswork/protein/ 2. Klaips CL、Jayaraj GG、Hartl FU。衰老和疾病中的细胞蛋白质稳态途径。J Cell Biol。2017;217(1):51-63。doi:10.1083/jcb.201709072 3. Ciechanover A。蛋白水解:从溶酶体到泛素和蛋白酶体。Nat Rev Mol Cell Biol。2005;6(1):79-87。doi:10.1038/nrm1552 4. Oprea TI、Bologa CG、Brunak S 等人。人类基因组中未探索的治疗机会。天然药物发现评论。2018;17(5):317-332。doi:10.1038/nrd.2018.14 5. Hopkins AL、Groom CR。可用药基因组。天然药物发现评论。2002;1(9):727-730。doi:10.1038/nrd892 6. Collins I、Wang H、Caldwell JJ、Chopra R。通过调节泛素-蛋白酶体途径进行靶向蛋白质降解的化学方法。Biochem J。2017;474(7):1127-1147。doi:10.1042/BCJ20160762 7. Ito T、Ando H、Suzuki T 等人。确定沙利度胺致畸性的主要靶点。Science。 2010;327(5971):1345-1350。doi:10.1126/science.1177319 8. Krönke J、Udeshi ND、Narla A 等。来那度胺可导致多发性骨髓瘤细胞中 IKZF1 和 IKZF3 选择性降解。Science。2014;343(6168):301-305。doi:10.1126/science.1244851 9. Lu G、Middleton RE、Sun H 等。骨髓瘤药物来那度胺可促进 cereblon 依赖性 Ikaros 蛋白破坏。Science。2014;343(6168):305-309。 doi:10.1126/science.1244917 10. Gandhi AK、Kang J、Havens CG 等。免疫调节剂来那度胺和泊马度胺通过调节 E3 泛素连接酶复合物 CRL4(CRBN.) 诱导 T 细胞阻遏物 Ikaros 和 Aiolos 降解,从而共刺激 T 细胞。Br J Haematol。2014;164(6):811-821。doi:10.1111/bjh.12708 11. Chamberlain PP、Lopez-Girona A、Miller K 等。人类 Cereblon–DDB1–来那度胺复合物的结构揭示了对沙利度胺类似物反应的基础。Nat Struct Mol Biol。2014;21(9):803-809。 doi:10.1038/nsmb.2874 12. Ito T, Handa H. Cereblon 及其下游底物作为免疫调节药物的分子靶点。Int J Hematol。2016;104(3):293-299。doi:10.1007/s12185-016-2073-4 13. Matyskiela ME, Lu G, Ito T 等人。一种新型 cereblon 调节剂将 GSPT1 募集到 CRL4 CRBN 泛素连接酶中。Nature。2016;535(7611):252-257。doi:10.1038/nature18611 14. Chamberlain PP, Cathers BE。Cereblon 调节剂:低分子量蛋白质降解诱导剂。Drug Discov Today Technol。 2019;31:29-34。doi:10.1016/j.ddtec.2019.02.004 15. Baek K、Schulman BA。分子胶概念固化。Nat Chem Biol。2020;16(1):2-3。doi:10.1038/s41589-019-0414-3 16. Scheepstra M、Hekking KFW、van Hijfte L、Folmer RHA。药物发现中用于蛋白质降解的双价配体。Comput Struct Biotechnol J。2019;17:160-176。doi:10.1016/j.csbj.2019.01.006

糖酵解和糖体中的蛋白质易位...
摘要。锥虫会引起被忽视的热带疾病,本综述讨论了针对糖酵解和糖体内部蛋白质易位作为治疗这些感染的策略的潜力。不同的研究表明,糖酵解是克氏锥虫、布氏锥虫和利什曼原虫等寄生虫的主要能量来源,它们的糖酵解酶与人类糖酵解酶有很大不同,为选择性药物开发提供了机会。抑制糖酵解可导致寄生虫大量死亡,因为即使部分阻断该途径也会破坏三磷酸腺苷的产生,而三磷酸腺苷对于寄生虫的生存至关重要。本综述还研究了跨糖体膜的蛋白质易位机制,特别是过氧化物酶的关键作用;糖体蛋白的错误定位会对寄生虫的生存产生不利影响。了解蛋白质输入的机制和糖体酶的独特特性可以促进针对这些特定目标的合理药物设计。总体而言,本综述强调需要创新的治疗方法来有效应对锥虫病带来的挑战,并主张进一步研究这些寄生虫的代谢脆弱性,以开发有针对性的有效治疗方法。

神经科学中的靶向蛋白质降解
本篇综述探讨了靶向蛋白质降解 (TPD) 这一新兴领域及其在神经科学和临床开发中的有希望的应用。TPD 提供了调节蛋白质水平的创新策略,代表了小分子药物发现和治疗干预的范式转变。重要的是,小分子蛋白质降解剂专门针对中枢神经系统细胞并去除致病蛋白质,而不存在基因组和基于抗体的模式的药物输送挑战。在这里,我们回顾了 TPD 技术的最新进展,重点介绍了具有邻近诱导降解事件驱动和迭代药理学的蛋白水解靶向嵌合体 (PROTAC) 蛋白质降解剂分子,提供了在神经科学研究中的应用,并讨论了将 TPD 转化为临床环境的巨大潜力。
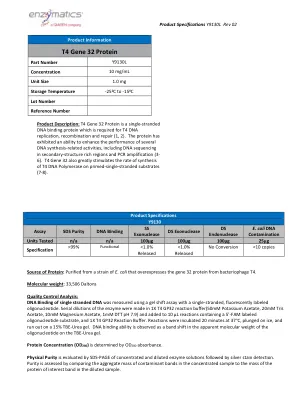
Qiagen®多路复用PCR加套件T4基因32蛋白
蛋白质的来源:从大肠杆菌的菌株中纯化,该菌株过表达了噬菌体T4的基因32蛋白。分子量:33,506 Daltons质量控制分析:使用带有单链,荧光标记的寡核苷酸标记的凝胶移位测定法测量了单链DNA的DNA结合。Serial dilutions of the enzyme were made in 1X T4 GP32 reaction buffer(50mM Potassium Acetate, 20mM Tris Acetate, 10mM Magnesium Acetate, 1mM DTT pH 7.9) and added to 10 µL reactions containing a 5'-FAM labeled oligonucleotide substrate, and 1X T4 GP32 Reaction Buffer.在37°C下孵育20分钟,将其浸入冰上,并在15%的TBE-TEREA凝胶上耗尽。DNA结合能力被视为在TBE-rea凝胶上寡核苷酸的表观分子量中的带移。蛋白浓度(OD 280)由OD 280吸光度确定。物理纯度,然后进行银色染色检测。通过比较浓缩样品中污染物带的聚集质量与稀释样品中蛋白蛋白蛋白带的质量来评估纯度。

蛋白质工程锦标赛的结果
蛋白质工程的巨大挑战是开发计算模型来表征和生成任意功能的蛋白质序列。进展受到缺乏1)为基准机会的限制,2)大蛋白质功能数据集和3)获得实验蛋白质表征。我们介绍了蛋白质工程锦标赛,这是一项旨在促进蛋白质工程中计算方法的开发和评估的全面竞争。该比赛由一个硅圆形组成,可预测蛋白质序列的生物物理特性,然后是一个体外圆形,其中使用自动方法设计,表达和表征新颖的蛋白质序列。完成后,所有数据集,实验协议和方法都可以公开可用。我们详细介绍了一场试点锦标赛的结构和结果,其中涉及七个蛋白质设计团队,由六个多目标数据集提供动力,并由我们的合作伙伴,国际风味和香水进行实验表征。即将举行的蛋白质工程锦标赛旨在动员科学界对该领域的进步进行透明评估。