XiaoMi-AI文件搜索系统
World File Search SystemProtein

DNA,RNA和蛋白质合成
在1940年代初期,奥斯瓦尔德·艾弗里(Oswald Avery)和他的同事着手测试格里菲斯(Griffith)实验中的转化剂是蛋白质,RNA还是DNA。科学家使用酶分别破坏了热杀死的S细胞中的三个分子中的每个分子。他们使用蛋白酶在第一个实验中使用蛋白酶破坏了热杀死细胞中的蛋白质,一种称为RNase的酶在第二个实验中破坏RNA,而一种称为DNase的酶在第三个实验中销毁DNA。然后,他们分别将热杀死的S细胞的三个实验批次与活细胞混合在一起,并用混合物将小鼠注入。缺少蛋白质和RNA的细胞能够将R细胞转化为S细胞并杀死小鼠。然而,缺少DNA的细胞没有将R细胞转化为S细胞,小鼠存活。
凤凰热启动TAQ DNA聚合酶
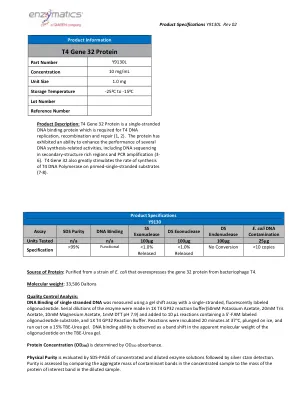
蛋白质的来源:从大肠杆菌的菌株中纯化,该菌株过表达了噬菌体T4的基因32蛋白。分子量:33,506 Daltons质量控制分析:使用带有单链,荧光标记的寡核苷酸标记的凝胶移位测定法测量了单链DNA的DNA结合。Serial dilutions of the enzyme were made in 1X T4 GP32 reaction buffer(50mM Potassium Acetate, 20mM Tris Acetate, 10mM Magnesium Acetate, 1mM DTT pH 7.9) and added to 10 µL reactions containing a 5'-FAM labeled oligonucleotide substrate, and 1X T4 GP32 Reaction Buffer.在37°C下孵育20分钟,将其浸入冰上,并在15%的TBE-TEREA凝胶上耗尽。DNA结合能力被视为在TBE-rea凝胶上寡核苷酸的表观分子量中的带移。蛋白浓度(OD 280)由OD 280吸光度确定。物理纯度,然后进行银色染色检测。通过比较浓缩样品中污染物带的聚集质量与稀释样品中蛋白蛋白蛋白带的质量来评估纯度。
肽和蛋白质治疗的成果
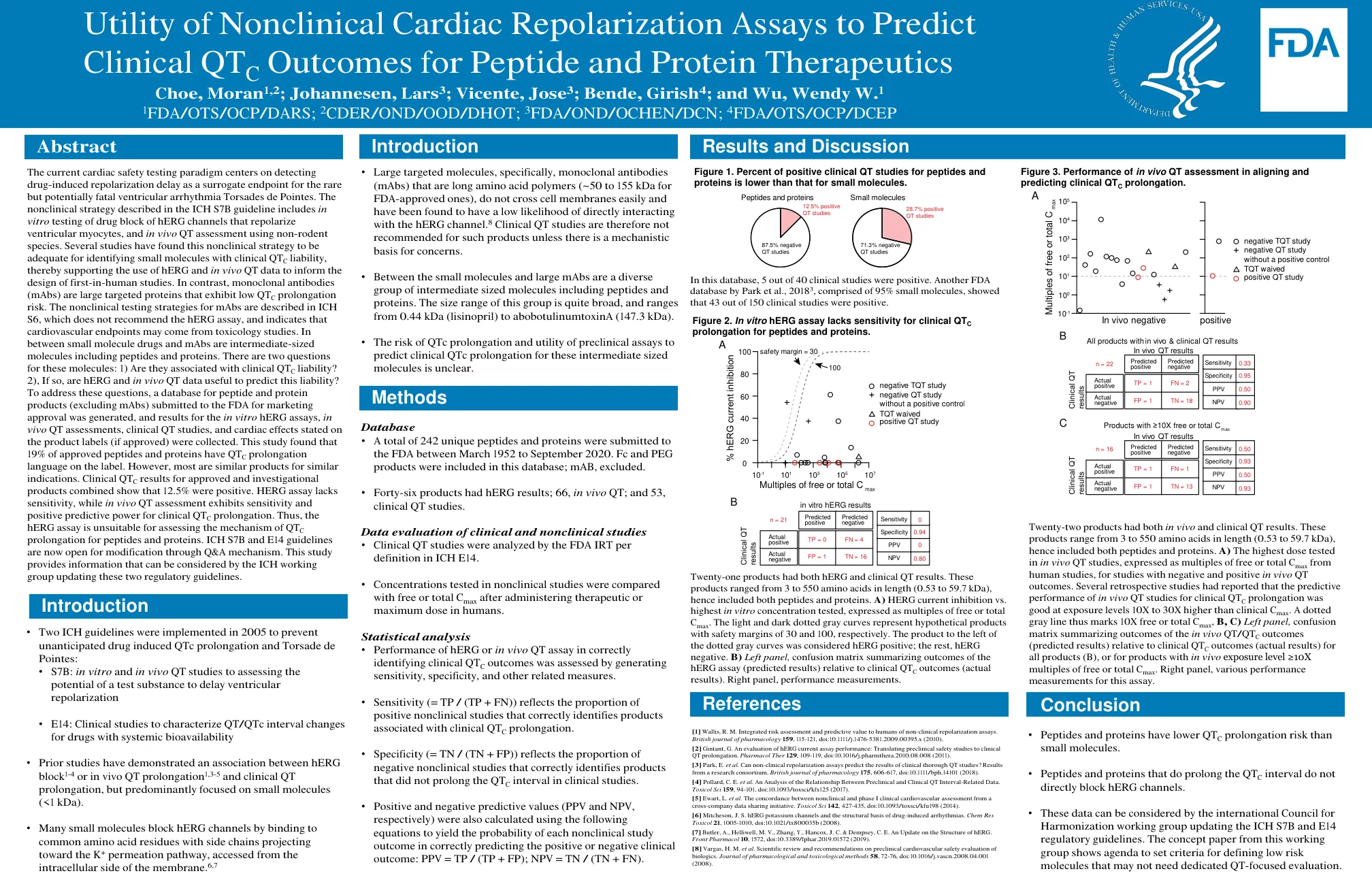
当前的心脏安全性测试范例以检测药物引起的复极延迟为中心,将其作为罕见但可能致命的室性心律失常尖端扭转型室性心动过速的替代终点。ICH S7B 指南中描述的非临床策略包括体外测试药物阻断 hERG 通道以复极心室肌细胞,以及使用非啮齿动物进行体内 QT 评估。多项研究发现,这种非临床策略足以识别具有临床 QT C 倾向的小分子,从而支持使用 hERG 和体内 QT 数据来指导首次人体研究的设计。相比之下,单克隆抗体 (mAb) 是表现出低 QT C 延长风险的大型靶向蛋白。ICH S6 中描述了 mAb 的非临床测试策略,它不推荐 hERG 检测,并表明心血管终点可能来自毒理学研究。小分子药物和 mAb 之间是中等大小的分子,包括肽和蛋白质。对于这些分子,有两个问题:1) 它们是否与临床 QT C 倾向有关?2) 如果是,hERG 和体内 QT 数据是否有助于预测这种倾向?为了回答这些问题,我们生成了提交给 FDA 以获得上市批准的肽和蛋白质产品(不包括 mAb)的数据库,并收集了体外 hERG 检测、体内 QT 评估、临床 QT 研究和产品标签上注明的心脏影响(如果已获批准)的结果。这项研究发现,19% 的获批肽和蛋白质在标签上有 QT C 延长的语言。但是,大多数是用于类似适应症的类似产品。已获批产品和研究产品的临床 QT C 结果综合显示 12.5% 为阳性。HERG 检测缺乏敏感性,而体内 QT 评估对临床 QT C 延长表现出敏感性和阳性预测力。因此,hERG 检测不适合评估肽和蛋白质的 QT C 延长机制。 ICH S7B 和 E14 指南现已开放通过问答机制进行修改。本研究提供的信息可供 ICH 工作组在更新这两项监管指南时参考。

基于蛋白质结构域的药物预测
虚拟筛选等预测方法已用于药物研发,目的是减少开发时间和成本。当前的机器学习和基于网络的方法存在与泛化、可用性或模型可解释性相关的问题,特别是由于目标蛋白的结构/功能的复杂性以及系统训练数据集的偏差。在这里,我们提出了一种新方法“DRUIDom”(DRUg 相互作用域预测),利用蛋白质的结构域模块化来识别药物候选化合物和靶标之间的生物相互作用,以克服与当前方法相关的问题。DRUIDom 由两个方法步骤组成。首先,将配体/化合物统计地映射到其靶蛋白的结构域,目的是识别它们的相互作用。这样,包含相同映射域或域对的其他蛋白质就成为相应化合物的新候选靶标。接下来,根据分子相似性对百万级小分子化合物数据集(包括上一步中映射到域的化合物)进行聚类,并将它们的域关联传播到同一聚类内的其他化合物。从公共数据库获得的经过实验验证的生物活性数据点经过精心筛选,构建活性/相互作用和非活性/非相互作用药物/化合物-靶标对的数据集(约 290 万个数据点),并用作计算化合物-域映射参数的训练数据,从而得到 250 个域和 8,165 种化合物之间的 27,032 个高置信度关联,最终输出约 500 万个新的化合物-蛋白质相互作用。通过对预测靶向 LIM-激酶蛋白的化合物进行合成和生物活性分析,对 DRUIdom 进行了实验验证,LIM-激酶蛋白在通过肌动蛋白丝动力学调节细胞运动、细胞周期进程和分化方面发挥关键作用。我们发现 LIMK-inhibitor-2 及其衍生物通过抑制 LIMK 磷酸化和下游蛋白肌动蛋白丝切蛋白,显著阻止癌细胞迁移。

基于蛋白质结构域的药物预测
虚拟筛选等预测方法已用于药物研发,目的是减少开发时间和成本。当前的机器学习和基于网络的方法存在与泛化、可用性或模型可解释性相关的问题,特别是由于目标蛋白的结构/功能的复杂性以及系统训练数据集的偏差。在这里,我们提出了一种新方法“DRUIDom”(DRUg 相互作用域预测),利用蛋白质的结构域模块化来识别药物候选化合物和靶标之间的生物相互作用,以克服与当前方法相关的问题。DRUIDom 由两个方法步骤组成。首先,将配体/化合物统计地映射到其靶蛋白的结构域,目的是识别它们的相互作用。这样,包含相同映射域或域对的其他蛋白质就成为相应化合物的新候选靶标。接下来,根据分子相似性对百万级小分子化合物数据集(包括上一步中映射到域的化合物)进行聚类,并将它们的域关联传播到同一聚类内的其他化合物。从公共数据库获得的经过实验验证的生物活性数据点经过精心筛选,构建活性/相互作用和非活性/非相互作用药物/化合物-靶标对的数据集(约 290 万个数据点),并用作计算化合物-域映射参数的训练数据,从而得到 250 个域和 8,165 种化合物之间的 27,032 个高置信度关联,最终输出约 500 万个新的化合物-蛋白质相互作用。通过对预测靶向 LIM-激酶蛋白的化合物进行合成和生物活性分析,对 DRUIdom 进行了实验验证,LIM-激酶蛋白在通过肌动蛋白丝动力学调节细胞运动、细胞周期进程和分化方面发挥关键作用。我们发现 LIMK-inhibitor-2 及其衍生物通过抑制 LIMK 磷酸化和下游蛋白肌动蛋白丝切蛋白,显著阻止癌细胞迁移。

XPack™外泌体蛋白质工程
SBI 优化了一种特定的肽序列,该序列可将蛋白质靶向到外泌体内膜,从而使融合蛋白能够被包装到外泌体中分泌。该“XPack”肽序列已被整合到 XPack 克隆和表达慢病毒载体中,从而允许将报告蛋白或任何感兴趣的蛋白质加载到生产细胞系内的外泌体中。然后可以分离这些工程外泌体并将其递送到目标受体细胞中。此外,已经生成了稳定的 XPack 细胞系,这些细胞系可作为装载报告蛋白货物的外泌体的恒定来源,从而能够在人类细胞和体内鼠模型中追踪外泌体的动态。来自这些稳定细胞系的纯化、即用型外泌体也可作为综合 XPack 系统的一部分使用。

活化蛋白 (FAP) PET
Thomas A. Hope,医学博士,1-3 Jeremie Calais,医学博士,哲学博士,4-5 Ajit H. Goenka,医学博士,6 Uwe Haberkorn,医学博士,7 4 Mark Konijnenberg,哲学博士,8 Jonathan McConathy,医学博士,9 Daniela E. Oprea-Lager,医学博士,10 Laura 5 Trimnal,3 Elcin Zan,医学博士,11 Ken Herrmann,医学博士,12-13 Christophe M. Deroose,医学博士 14-15 6

目标蛋白质降解概况
截至 2024 年 9 月,我们组建了一个包含 256 种靶向蛋白质降解剂 (TPD) 的综合数据库,涵盖所有研究、临床前、临床开发和上市资产。核心来源。我们使用 EvaluatePharma 数据库汇编了我们的初始资产集。为了识别相关资产,我们根据 EvaluatePharma 整体研发管线数据库的“作用机制”或“药物类别”列中的关键词(“降解剂”、“PROteolysis Targeting Chimera”、“PROTAC”、“BiDAC”、“免疫调节药物”、“IMiD”、“Cereblon E3 连接酶调节药物”、“CELMoD”、“SERD”、“分子胶”、变体)对数据库进行了过滤。验证。我们手动将生成的数据库与公司网站进行交叉检查,以将该机制归类为 TPD,结果删除了约 20 种被错误归类为 TPD(例如抑制剂、激动剂、抑制剂)的资产。为了验证我们的列表是否是最新的,我们使用相同的 TPD 关键词扫描了最近的新闻稿、会议报告、学术出版物和公司网站。这次扫描增加了两个最近进入临床的资产,并删除了六个最近已停止的资产。鉴于 EvaluatePharma 的捕获偏差,我们预计研究、临床前和美国/欧盟以外资产的覆盖率会较低。技术。对于归类为 TPD 的资产,我们根据技术将其分为当前一代、下一代和未指定。当前的技术有:分子胶(包括 IMiD 和 CELMoD);异双功能降解剂(包括直接募集普遍表达的 E3 连接酶的小分子异双功能降解剂,例如 PROTAC);选择性雌激素受体降解剂 (SERD)。正在研发的下一代技术有:降解剂-抗体偶联物 (DAC);细胞外蛋白的分子降解剂 (MoDE);伴侣介导 (CHAMP);自噬 (AUTAC)。请注意,根据分子结构和机制,雌激素受体降解剂被归类为 SERD 和异双功能降解剂;例如,elacestrant 被归类为 SERD,而 vepdegestrant 被归类为异双功能降解剂。另请注意,鉴于计划启动与自噬研究状态之间的比较,三种结构未公开的异双功能“自噬刺激剂”被归类为 AUTAC,但可能是类似的自噬技术(例如 ATTEC)。开发阶段。“已批准”资产目前已获批准。“临床”资产目前处于 I-III 期临床试验阶段,使用 ClinicalTrials.gov 和/或公司网站确定。“研究”资产目前正在积极研究或临床前开发中,使用新闻稿和/或公司网站确认。治疗领域。对于上市资产,我们使用批准新闻稿和药物说明书手动确认治疗领域。对于临床资产,我们使用 ClinicalTrials.gov 和/或公司网站手动确认治疗领域;如果某项资产存在多项临床试验,则使用最先进的试验来确定治疗领域。对于研究和临床前资产,我们从 EvaluatePharma 数据库中提取治疗领域。治疗领域被合并为资产数量最多的三个类别(癌症、神经病学和免疫学),然后是“其他”,包括泌尿道、感染、呼吸道、皮肤、糖尿病、胃肠道、肌肉骨骼、肝脏、血液、心血管、泌尿道和其他(每种有 1-5 个资产)。目标。对于已上市资产,我们使用批准新闻稿和药品说明书手动确认目标蛋白。对于临床资产,我们使用 ClinicalTrials.gov 和/或公司网站手动确认目标蛋白。对于研究和临床前资产,目标蛋白是根据“作用机制”和“药物类别”字段中的关键词的人工检查来确定的,例如 SMARCA2 降解剂、α-突触核蛋白 (SNCA) 降解剂,并辅以公司网站、新闻稿和 Citeline。

