XiaoMi-AI文件搜索系统
World File Search SystemPy

肯塔基州 2022 年经济分析
本劳动力产品由美国劳工部就业和培训管理局的拨款资助。该产品由受助人创建,并不一定反映美国劳工部的官方立场。美国劳工部对此类信息不作任何明示或暗示的担保、保证或保证,包括链接站点上的任何信息,包括但不限于信息的准确性或完整性、及时性、实用性、充分性、持续可用性或所有权。本产品的版权归创建它的机构所有。组织内部使用和/或个人用于非商业目的是允许的。所有其他用途均需事先获得版权所有者的授权。联邦政府保留复制、出版或以其他方式使用,并授权他人用于联邦目的的已付费、非独占和不可撤销的许可,并保留以下权利:i) 根据拨款开发的所有产品的版权,包括拨款或分拨款下的分拨款或合同;以及 ii) 受赠人、次级受赠人或承包商根据奖励购买的任何版权权利(包括但不限于课程、培训模型、技术援助产品和任何相关材料)。此类使用包括但不限于以任何方式(电子或其他方式)修改和分发此类产品的权利。联邦资金不得用于支付使用受版权保护作品的任何版税或许可费,或通过购买作品版权获得的费用,如果该部门拥有此类作品的许可或免费使用权,但它们可用于支付获取副本的费用,但仅限于开发商/卖家的复制和运输费用。如果收入是通过销售使用赠款资金开发的产品(包括知识产权)产生的,则这些收入属于计划收入。计划收入必须按照本赠款奖励和 2 CFR 200.307 的规定使用。

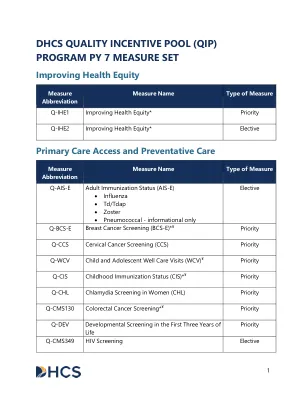
2025 年支付和归因方法
ACM 门诊联合管理 ACO 责任医疗组织 ACO REACH 责任医疗组织实现公平、可及性和社区健康 ADI 区域贫困指数 AHEAD 推进全额付款人健康公平方法和发展 AHRQ 医疗保健研究与质量机构 BAL 受益人证明清单 CC 病情类别 CCA 协作护理安排 CCM 慢性病管理 CCN CMS 认证编号 CG-CAHPS 临床医生和团体消费者对医疗保健提供者和系统的评估® CI 持续改进 CMS 医疗保险和医疗补助服务中心 CPT 现行程序术语 CQM 临床质量测量 DXG 诊断组 E&M 评估和管理 eCQM 电子临床质量测量 ED 急诊科 EDU 急诊科利用率 ESP 增强服务付款 ESRD 终末期肾病 FFS 按服务收费 FQHC 联邦合格医疗中心 GAF 地理调整因素 HCC 分层病情类别 HCPCS 医疗保健通用程序编码系统 HEDIS 医疗保健有效性数据和信息集 HIPAA 健康保险流通与责任法案 HPSA 卫生专业人员短缺领域 IPC 跨专业咨询 IT 信息技术 ITU 印第安人健康服务、部落和城市印第安人提供者 LIS 低收入补贴 MCP 使护理成为初级 MEC MCP 电子咨询 MIPS 择优激励支付系统 NCQA 国家质量保证委员会 NPI 国家提供者标识符 PA 参与协议 PBPM 每位受益人每月 PCF 初级保健优先 PCPCM 以人为本的初级保健措施

肯塔基州 2020 年经济分析
本劳动力产品由美国劳工部就业和培训管理局拨款资助。本产品由受助人创建,并不一定反映美国劳工部的官方立场。美国劳工部不对此类信息(包括链接网站上的任何信息)提供任何明示或暗示的保证、担保或担保,包括但不限于信息的准确性、完整性、及时性、实用性、充分性、持续可用性或所有权。本产品的版权归创建它的机构所有。组织内部使用和/或个人出于非商业目的的个人使用均允许。所有其他用途均需事先获得版权所有者的授权。

肯塔基州 2021 年经济分析
2021 年计划年度 (PY) 肯塔基州经济分析报告由肯塔基州统计中心 (KYSTATS) 劳动力情报处制作,旨在详细分析该州及其十个地方劳动力区 (LWA) 的经济和劳动力。本报告根据美国劳工部、就业和培训管理局 (ETA) 发布的培训和就业指导信 (TEGL) 第 1-21 号编写。该分析对肯塔基州的劳动力市场状况和经济进行了深入审查,包括人口、人口统计、劳动力以及工业和职业就业趋势和活动。该报告还将该州的经济成果与整个国家进行比较。本报告主要关注 2011 年至 2021 年。职业预测估计涵盖 2019 年至 2029 年。为了更好地了解肯塔基州经济的多样性,本报告还提供了该州十个 LWA 的人口统计和经济信息,如下图 1 所示。LWA 的指定是 2014 年《劳动力创新和机会法案》 (WIOA) 规定的。与组成它们的县一样,这十个 LWA 在失业率、职业就业和增长、人口和教育程度方面可能存在很大差异。一般而言,与较为农村的 LWA 相比,规模较大、较为城市化的 LWA 具有较高的教育水平、较高的就业增长率和较低的失业率。本报告使用了多个数据来源。整体经济的衡量指标包括国内生产总值 (GDP) 和个人收入,这些数据来自美国经济分析局 (BEA)。人口和人口预测来自路易斯维尔大学的肯塔基州数据中心。其他人口统计信息来自美国人口普查局的 2020 年人口普查和美国社区调查 (ACS)。ACS 5 年估计值(2016-2020 年)提供了县级经济和人口特征的信息。所有 120 个县的数据被汇总以提供 LWA 级别的估计值。劳动力数据来自劳工统计局 (BLS) 计划。有关美国劳动力的信息来自当前人口调查 (CPS)。肯塔基州和十个 LWA 的其他劳动力数据来自 KYSTATS 劳动力情报处管理的当地失业统计 (LAUS) 计划。季度就业和工资普查 (QCEW) 计划的数据用于衡量所涵盖行业的就业和工资随时间的增长。职业就业和工资统计 (OEWS) 计划和 KYSTATS 提供了 700 多种详细职业的职业估算。

ARA® XTREME PY 2100 美国
ARA ® XTREME PY 2100 US 是一种粘度极低、功能性强、纯度高的胺基树脂,具有相对良好的储存稳定性。它固化速度非常快,可生产出具有极高热变形温度的产品。ARA ® XTREME PY 2100 US 是一种特别有效的树脂,适用于各种配方应用,包括粘合剂、层压系统等。它可以用作粘度调节剂,也可以与慢反应性树脂一起使用以提高其固化速度;但是,由于其快速固化特性,在选择固化剂和固化条件时必须谨慎。即使是适量的树脂,在与脂肪胺固化时,也会产生足够的放热,导致烧焦和冒烟。如果芳香胺硬化系统在过高的温度下凝胶化,或者单独使用或与芳香族硬化剂结合使用催化剂(例如三氟化硼单乙胺),也会出现这种情况。 ARA ® XTREME PY 2100 US 是对氨基苯酚的三缩水甘油酯,其化学结构如下所示。

2024 年度行动计划草案
坦帕市是美国住房和城市发展部 (HUD) 指定的授权城市。因此,该市获得了社区发展综合拨款 (CDBG)、HOME 投资合作伙伴计划 (HOME)、紧急解决方案拨款 (ESG) 和艾滋病患者住房机会 (HOPWA) 拨款。该市的住房和社区发展部 (HCD) 管理联邦 HUD 授权拨款资金,并致力于为坦帕的低收入至中等收入 (LMI) 个人和有特殊需要的人群提供安全、体面和负担得起的住房以及合适的生活环境。与其他重要市政府部门、非营利性利益相关者组织和公众公民的合作使 HCD 能够实现其满足社区需求的目标。

PY 536 - 量子计算 2019 年秋季
学术行为 您可以与其他学生合作完成家庭作业、准备口头报告以及进行期末论文研究。但是,您提交的家庭作业和期末论文的撰写以及您展示的幻灯片应代表您个人的努力。所有学生都有责任了解和理解 CAS 学术行为准则的规定。

关岛防寒保暖州计划 2025 年
一、概述 防寒保暖援助计划(WAP)由 1976 年《能源保护与生产法》第四章创立,旨在帮助缺乏资源投资于能源效率的低收入家庭。这项联邦法令授权美国能源部(USDOE)管理 WAP(42 USC § 6861 et seq.)。WAP 为符合条件的低收入家庭免费提供一定限额的财政援助,以提供由联邦规则确定的全屋防寒保暖服务,从而最大限度地节省能源和资金。2007 年《能源独立与安全法》重新授权 WAP,扩大了“州”定义中美国任何领土或属地作为符合条件的受助者的范围。在 2009 项目年度(PY),领土首次被纳入 WAP 拨款的接受范围。自 2009 年以来,关岛能源办公室 (GEO) 一直是实施关岛领地 WAP 的资助接受者。为了每年根据 WAP 获得联邦资金,各州和领地需要根据 10 CFR 440 中的 WAP 法规、2 CFR 200 中的美国能源部财政援助规则以及美国能源部防寒保暖计划通知中包含的所有后续指导制定自己的 WAP 州计划。所有美国能源部规则和指导都可以在其网站上找到。在最终确定并提交资助申请文件中的必要信息之前,现将关岛 2025 年计划年度的防寒保暖州计划提交给美国能源部,征求公众意见。一旦获得联邦资金,GEO 将从 2025 年 4 月 1 日至 2026 年 3 月 31 日开始为符合条件的低收入家庭实施 WAP 服务,以减少家庭能源支出,同时确保和增强家庭的健康和安全。II。联邦援助 联邦援助资金分配基于美国国会在向各州和领地分配资金时制定的 WAP 公式。关岛有资格获得 292,187 美元的联邦资金,用于在 2025 财年(从 2025 年 4 月 1 日开始到 2026 年 3 月 31 日)为符合条件的收入家庭实施 WAP 服务,该资金将基于《防寒保暖计划通知》25-2 的分配。WAP 拨款是一个为期 3 年的拨款周期。在发布最终拨款后,将对拨款州计划进行适当调整。

sjvac rpu py 2025-28区域计划
I.随着2014年《劳动力创新与机会法案》(WIOA)的通过,国会在如何满足经济和劳动力市场需求方面朝着新的方向发展了国家劳动力系统。虽然在过去的四十年中建立当地劳动力地区始终考虑了当地劳动力市场,但Wioa认识到经济往往是区域性的,很容易溢出在商业发现无关紧要的管辖权边界上。WIOA对区域经济的认可引起了加利福尼亚建立区域规划部门(RPU)的建立,该部门代表了当地劳动力地区的群体,这些劳动力地区共同开发反映了企业和劳动力地区经济需求的战略。根据联邦和州的指导,圣华金河谷和关联县(SJVAC)RPU制定了这项为期四年的区域计划,以指导整个计划年度的战略计划(PY)2025-28,该计划涵盖2025年7月1日至2025年6月30日至2029年6月30日。A.劳动力创新与机会法案与两党多数席位通过,WIOA旨在帮助求职者获得就业,教育,培训和支持服务,以在劳动力市场中取得成功,并将雇主与他们在全球经济中竞争的熟练工人相匹配。WIOA代表了联邦劳动力立法的最新版本,向各州和当地提供资金来管理和运营劳动力发展计划。WIOA之前是《职业培训合作法》(1982年至2000年的活动)和《劳动力投资法》(2000年至2015年的活跃)。