XiaoMi-AI文件搜索系统
World File Search SystemPyrosequencing

从其成分Meju和太阳盐中对细菌迁移到Doenjang的培养依赖性和非依赖性研究
我们使用基于培养物和16S rRNA基因基因培养的非依赖性技术(总DNA的pycecing)从其成分(MEJU和太阳盐)中确定细菌迁移到Doenjang。焦磷酸测序结果表明,Meju但没有太阳盐的细菌群落显着影响Doenjang社区的细菌群落。基于培养的焦磷酸测序分析产生了相似的结果。这些结果表明,doenjang中的大多数主要细菌物种是从Meju而不是太阳盐迁移的。因此,我们认为本研究是使用依赖培养和独立的方法的发酵大豆的细菌沟通最全面的比较之一。更重要的是,细菌16S rRNA的V3和V4区域的焦磷酸测序没有区分阿米洛基法西氏芽孢杆菌,b。siamensis,b。velezensis以及粪肠球菌和E之间。Hirae。Hirae。

IQ亚型,大脑体积和免疫标记
当前的化学测试策略的检测能力有限,其检测非生物毒性致癌物(NGTXC)的能力受到限制。表观遗传异常在癌变期间发生,无论分子启动事件是否与遗传毒性(GTXC)或NGTXC事件有关;因此,可以利用表观遗传标记来开发新的方法方法,以改善两种类型的致癌物的检测。这项研究使用叙利亚仓鼠胎儿细胞来建立致癌物诱导的DNA甲基化从原代细胞变化,直到衰老 - 衰老,这是必不可少的致癌步骤。将暴露于溶剂对照7天的细胞与幼稚的原发性培养物进行了比较,与苯并[a] pyrene暴露了7天的细胞,以及随后转化阶段的细胞:正常菌落,形态转化的菌落,衰老,衰老,衰老,bypass和持续的扩散。DNA甲基化变化通过降低的代表性亚硫酸盐测序在第7天最少。在细胞衰老过程中产生了深刻的DNA甲基化变化,其中一些早期差异甲基化区域(DMR)通过最终的持续性促进阶段保留。通过Pyrosequencing验证了一组这些DMR(例如POU4F1,AIFM3,B3GALNT2,BHLHE22,GJA8,KLF17和L1L),并通过Pyrosequencing验证,并在从不同的实验室中获得的多个克隆中证实了它们的可重复性。这些DNA甲基化变化可以用作生物标志物,以增强对细胞转化的客观性和机械理解,并可用于预测衰老 - 肿瘤和化学致癌性。

课程描述植物学
基因组学分类,原核基因组的结构和组织。细菌基因的转录调节剂。细菌基因组中的可转座遗传元素。细菌操纵子和操纵片化的演变。岛屿和致病性和抗性的片段。真核基因组的结构和组织。重复和转座元素及其对基因组的影响。染色体中的端粒和亚电体区域。CpG甲基化和基因沉默。 酵母 - 两种杂交系统。 cDNA微阵列。 线粒体基因组的进化和结构。 基因组测序:整个shot弹枪基因组测序。 测序技术:Sanger毛细血管测序,Roche 454(焦磷酸测序),Illumina/Solexa,固体系统。 测序技术的优缺点。 Maxam-Gilbert测序。 ORF和启动子预测。 内含子和外显子预测。 基因注释。 主要基因组数据库。CpG甲基化和基因沉默。酵母 - 两种杂交系统。cDNA微阵列。线粒体基因组的进化和结构。基因组测序:整个shot弹枪基因组测序。测序技术:Sanger毛细血管测序,Roche 454(焦磷酸测序),Illumina/Solexa,固体系统。测序技术的优缺点。Maxam-Gilbert测序。ORF和启动子预测。 内含子和外显子预测。 基因注释。 主要基因组数据库。ORF和启动子预测。内含子和外显子预测。基因注释。主要基因组数据库。

卷∙17∙编号∙4∙2024 页码∙1423 描述性...
摘要 本研究旨在调查 CBCT 分析和 DNA 甲基化测量通过同一颗拔除牙齿估计人类年龄的潜力。对印度尼西亚帕查贾兰大学牙科医院的三颗拔除的下颌前磨牙进行了横断面方法的描述性研究设计。使用 ITK-SNAP 测量牙髓和牙齿体积进行 CBCT 分析,并使用组内相关系数 (ICC) 进行可靠性测试。同时,使用焦磷酸测序分析对 ELOVL2 基因进行 DNA 甲基化测量。使用平均绝对误差 (MAE) 和均方根误差 (RMSE) 量化每个样本的估计年龄和实际年龄之间的差异。在 CBCT 分析中,MAE 范围为 0.44 至 3.97 岁,RMSE 范围为 0.52 至 4.01 岁。至于 DNA 分析,MAE 为 1.37 岁,RMSE 为 1.67 岁。 CBCT 分析和 DNA 甲基化测量均已证明能够根据同一颗拔除的牙齿估计人类年龄。在这项初步研究中,基于牙髓-牙齿体积比的 CBCT 分析估计的人类年龄比 DNA 甲基化水平测量更接近实际年龄。
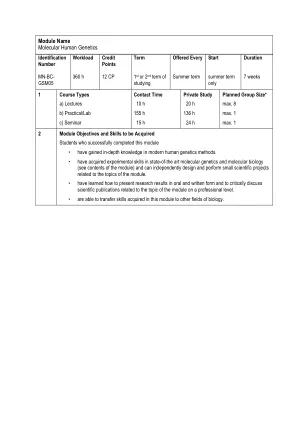
模块名称分子人遗传学
•人遗传疾病的分子基础(神经肌肉和神经退行性疾病,肾脏疾病,骨骼疾病和遗传性肿瘤倾向综合征)和罕见发育综合征的鉴定和表征。Subtopics: disease gene location (linkage studies), identification of disease genes (targeted (Panel) and whole exome sequencing using next generation sequencing), identification of underlying mutations, functional analysis of disease genes in vitro and in vivo, functional analysis of the disease relevant protein complexes • Identification of disease modifying/protective factors • Therapeutic approaches (pharmacotherapy, epigenetic approaches, gene疗法)•分子遗传技术(PCR,测序,实时PCR,多态性标记的基因分型,RT-PCR,焦磷酸测序,Southern-Clotting等)•分析测序数据和突变,单倍型的构建,引物的构造,序列的组装和对齐等。•分子克隆(将PCR片段克隆到质粒中,质粒DNA的分离,转染); use of CRISPR/Cas-system • Cell culture technology (working with human and murine cell lines) • Working with inducible pluripotent stem cells (iPSC) and neuronal differentiation • Immunohistochemistry, fluorescence microscopy • Protein analysis and protein-interaction methods (Western blotting, co-immunoprecipitation of proteins, pull-down, chromatin-immunoprecipitations (ChIP) etc.)•分析敲除和转基因小鼠的解释性注释:上面的列表包括人类遗传学,CECAD,CMMC,CCG,CCG,表观基因组学和眼睛的实验免疫学的主题和技术。因此,参加该模块的每个学生都将面临其中的大部分子集。确切的内容将取决于学生和研究项目的研究项目。

微生物在法医学中的作用
pooja jk doi:https://doi.org/10.33545/27074447.2023.v5.i1a.59摘要人类微生物组提到了所有微观生命形式,例如细菌,病毒,病毒,藻类和饮食人体身体。法医微生物学涉及基于验尸间隔及其在身体不同部位的分布来鉴定微生物,这有助于个人鉴定,死亡确定原因,地理位置确定可能在哪里发现尸体和体液识别。微生物法医用于研究由微生物在性侵犯案件,生物犯罪或任何其他形式的刑事案件中引起的微生物和疾病的传播。分子生物学和遗传学的进步导致了分析仪器和技术的发展,有助于更好地分析微生物样品及其代谢产物。thanato-Microbiology是指驻留在人体表面上的微生物研究,这也是法医微生物学研究领域,主要有助于基于独特的微生物居住的独特的微生物来区分另一个人。关键字:法医学微生物,thanato-Microbiology,pyrosequencing简介微生物或简称微生物是最小的单细胞生物。他们既有用,又对人类有害。它们分为不同类型,例如细菌,病毒,真菌和原生动物。细菌是最丰富的微生物,通常被分为两种类型,即考古细菌和花生细菌。(Zachary等,2017)[5]。(Zachary等,2017)[5]。人们认为,人体内部和人体上的细菌比人体细胞多十倍(Turnbaugh等,2009)[1]。研究表明,微生物在法医后检查,自死亡确定以来的时间,通过分析体液中发现的微生物群的个人鉴定,地理位置的鉴定,基于人体中的微生物种群发生死亡可能发生的地理位置。微生物,例如梭状芽胞杆菌,乳酸杆菌,eggerthella和细菌,在下部胃肠道中大量发现,而链球菌,prevotella和veillonella在人体的上部胃肠道中广泛分布。在前阳光期间的口腔中发现了富公司,而在肿胀期间则发现了蛋白质(Hyde等,2013)。用于鉴定微生物的常见方法包括焦磷酸测序和脉冲场凝胶电泳。其他检测方法包括16/18S核糖体RNA(rRNA)基因,单核苷酸多态性,内部转录的垫片和整个基因组shot弹枪。这些基因组方法在法医科学中很有用,可以创建遗传特征和鉴定整个微生物群落。对于蛋白质合成所必需的70s和80s核糖体是由16s和18s RNA组成的,通常在分类门中保持高度保守,但存在具有种间多态性或突变的可变区域,可帮助您识别个体。用于分类学分析的DNA的其他区域是核糖体RNA基因之间的非编码区域,称为内部转录间隔物(ITS),例如16S和23S细菌和古细菌(Lafontaine和Tollervey,Tollervey,2001年)。这些区域的突变率很高,因为它们的生存不是必不可少的,因此可以在相似的物种上进行比较(Baldwin,1992)。Mortem Microbial社区和PMI与宿主相关后与宿主相关的微生物群落称为Thanato-Microbiome。

EZ-96 DNA甲基化 - 光泽Magprep
胞嘧啶甲基化是原核和真核生物的天然基础修饰,包括通过甲基转移酶酶将甲基添加到胞质嘧啶环的第五碳位置中(1)。在原核生物中,DNA甲基化提供了一种方法,可以通过限制性核酸内切酶保护宿主DNA免受消化的影响,这些核酸内切酶旨在消除外源DNA。DNA甲基化在基因表达的调节/控制中的较高真核生物中的功能(2)。哺乳动物中的大多数DNA甲基化发生在5'-CPG-3'二核苷酸中,尽管确实存在其他模式。发现哺乳动物基因组中所有5'-CpG-3'二核苷酸的所有5'-CpG-3'二核苷酸被发现是甲基化的,而剩下的20%的二十%的二十%二十分位于启动子或最初的基因外显子内。已经证明异常DNA甲基化是癌症中普遍存在的现象,可能是肿瘤发生期间发生的最早变化之一(3)。DNA甲基化也已显示在基因印记,胚胎发育,X染色体基因沉默和细胞周期调节中起着核心作用。能够有效,准确地检测和量化DNA甲基化的能力对于研究癌症,基因表达,遗传疾病以及生物学的许多其他重要方面至关重要。迄今为止,已经开发了许多方法来检测/量化DNA甲基化,包括:高性能毛细管电泳(4)和甲基化敏感的任意启动PCR(5)。但是,当今使用的最常见技术仍然依赖于亚硫酸盐转化率(6)。用硫酸硫酸氢盐处理DNA化学将非甲基化的胞嘧啶修饰为尿嘧啶,甲基化的胞嘧啶保持不变。转换后,可以使用所需的下游应用确定DNA的甲基化曲线。为了进行单个基因座分析,在亚硫酸盐转化率(即Bisulfite PCR)之后,通常会扩增感兴趣的区域,然后对pyrosequencing®进行测序或处理。甲基化检测的最新进展还允许使用包括基于阵列的方法在内的技术,减少表示甲基甲基甲基化(RRBS)和整个基因组Bisulfite测序(7)。

原始人的自然经济
焦磷酸测序:Roche模板由EMPCR 1制备,其中1-20万珠沉积在PTP井中。较小的珠,带有连接的硫酸酶和荧光素酶围绕模板珠。单个DNTP依次流过井,以预定的顺序分配。在掺入补体DNTP时,释放的PP I被转换为ATP,从荧光素蛋白到羟基二耐蛋白的氧化产生光。读取平均400个基础作为流程图。对于均聚物,重复多达六个核苷酸,添加的DNTP的数量与光信号成正比。插入是最常见的错误类型,其次是删除。通过连接测序:将约1亿个EMPCR的模板珠沉积在载玻片上。在退火时,添加了1,2个探针的库。适当的条件使选择性杂交和探针结扎到互补位置。1,2探针的第一个(y)和第二(z)位置被设计为审讯库,因此16个二核苷酸由四种染料编码。在四色成像之后,将带状的1,2探针化学裂解以产生5'-PO 4组(P)。杂交,连接,成像和裂解的循环又重复了六次。然后从模板中剥离扩展引物,并使用N – 1底漆进行第二个连接弹,该底漆将询问底座重置为左侧的一个位置。询问每个基础两倍,提高了颜色调用的准确性。随后发生了七个连接周期,然后再进行三个结扎弹。然后将35个数据位组成的字符串在色彩空间中编码,然后对准参考基因组以解码DNA序列。替换是最常见的错误类型。可逆终结器:DNA片段的Illumina Bridge放大是在载玻片的八个通道上随机分布的,高密度向前和反向引物共价附加到其上。固相扩增可从单个ssDNA模板产生约8000万个MC。将底漆退火到每个MC中模板的自由末端。聚合酶延伸,然后终止从四个RTs组中的DNA合成,每组用不同的染料标记。未合并的RT被洗净,通过四颜色成像进行基础识别,并通过化学裂解去除阻塞和染料组以允许下一个周期。给定MC的颜色图像提供了〜45个基础的读取。替换是最常见的错误类型。使用RTS进行单分子测序:Helicos数十亿个未夸大的ssDNA模板是用poly(da)尾巴制备的,这些尾巴与聚(DT)引物杂交,共同连接到载玻片上。对于一通测序,该引物 - 模板复合物就足够了。两通序测序涉及复制模板链,删除原始模板,并退火向表面(未显示)。与Illumina的RT不同,这四个Helicos RT用相同的染料标记,并以预定的顺序单独分配。融合事件导致荧光信号。使用单分子消除了Dephasing的问题,其中给定MC内的数千个复制模板不会有效地扩展其引物。删除是最常见的误差类型,可以通过提供约25个基本共识读取的两次测序可大大降低。的应用和挑战100篇论文描述了这些创新的成果。虽然改进继续,但读取长度限制,错误类型和频率显着影响组装策略。对于简短(<100个基本)读取平台,通过映射到参考基因组来指导组装。结合Sanger和Roche数据(100个基本读数)改善了从头组件2,并且随着焦磷酸测序读取长度的改进,使用混合Roche(250键读数)和Illumina数据进行了改善,已经描述了从头组装。最近使用Roche 4和Illumina平台报告了第一个个性化基因组测序项目。Roche,Illumina和AB平台在1,000个基因组项目中被用于生成人类遗传变异的详细图表以及人类微生物组项目,以将微生物组动态与人类健康相关联。应用不限于测序基因组。共识计数分析5最近出现了,从而实现了转录因子结合,mRNA剪接,DNA甲基化,小RNA,染色质结构和DNase超敏位点的全局分析。配对的测序方案。这些不仅对从头组件很重要,而且对于识别结构变化和映射mRNA剪接同工型。展望未来,太平洋生物科学,多佛系统(Polonator G.007),Visigen Biotechnologies,Lasergen,Inc。,Intelligent Bio-Symys,完整的基因组学和牛津Nanopore技术等公司的平台开发。
法医学中的下一代测序
法医学中的下一代测序:一个引物解决了其针对法医科学应用的下一代测序(NGS)。本书的第一部分提供了人类认同方法的历史,包括VNTR,RFLP,STR和SNP DNA键入。它讨论了针对人DNA键入的测序历史,包括Sanger测序,快照,pyrosequencing和下一代测序的原理。这些章节概述了使用常染色体,Y和X染色体STR和SNP使用MISEQ FGX和ION TORRENT系统,概述了人类DNA键入的forenspo foseq,forenseq,forenseq,precision ID,powerSeq和QIASEQ面板。作者概述了在准备使用NGS试剂盒的库之前执行的DNA提取和DNA定量中包含的步骤。本书的后半部分详细介绍了ForenseQ和Precision ID的实现,以扩大和标记目标以创建库,丰富目标,以附加索引和适配器,执行库纯化和归一化,填充库,并将样品加载到墨盒上以在乐器上执行排序。覆盖范围解决了Miseq FGX和ION厨师的操作,包括创建样本列表,执行洗涤步骤,执行NG,了解仪器中的Run反馈文件以及故障排除。forenseq和精密ID面板数据分析将解释,包括如何分析和解释NGS数据以及输出图和图表。本书以线粒体DNA(mtDNA)测序和SNP分析结束,包括异质问题。最终章节回顾了微生物DNA,NGS在体液分析中的法医应用以及未来应用的挑战和考虑。特征 - 使用传统和NGS DNA键入方法针对人类识别,靶向短串联重复(Strs) - 将技术及其应用于执法调查,身份以及祖先的单核苷酸多态性(SNP)(SNP),以进行研究领导,大规模灾难和祖先的学生 - 在NG的习惯中,以实践为准。在法医计划中研究DNA这是第一本为从业人员准备并在其实验室中实施这项新技术的书籍,以进行案例工作,并强调了如何在法庭上使用NGS结果的早期应用。这本书可用于上级本科生和研究生,并参加了专注于NGS概念的课程。读者有望对分子和细胞生物学和DNA分类有基本的理解。

自闭症和肠道微生物群的有影响力文章:书目资料概况和研究趋势
自闭症谱系障碍(ASD)是一种神经发育障碍,其特征在于社会交流和互动中的缺陷,以及重复和限制性行为模式的表现(APA,2022)。估计ASD的全球患病率估计约为1%,并且在各个国家 /地区随着时间的推移,患病率的估计值有所增加(Zeidan等,2022年)。患有ASD的人可能会有情感和行为问题,例如自我伤害,侵略性,发脾气和财产破坏(Jang等,2011)。他们经常有其他精神病,例如焦虑,抑郁和精神病(Dan等,2020)。ASD的经济成本巨大,其中包括医疗服务的成本,特殊教育,患有ASD的人的生产损失,护理人员生产力损失和暂息护理(Rogge and Janssen,2019年)。在美国,据报道,ASD的急诊室服务的平均年度支出为15,929美元,而非ASD的平均每年支出为2,598美元; ASD的门诊就诊的年度支出为4,375美元,而非ASD为824美元(Vohra等,2017)。在英国,据估计,与ASD的青少年需要额外的特殊教育或住宅教育的成本在6个月内售价10,507英镑(Barrett等,2015)。 据报道,肠道菌群的组成与ASD有关。 肠道微生物群具有非常多样的组成,由细菌以及真菌,病毒和原生物组成(Enaud等,2018)。 它与中枢神经系统有双向联系。 例如,最近的文献计量学在英国,据估计,与ASD的青少年需要额外的特殊教育或住宅教育的成本在6个月内售价10,507英镑(Barrett等,2015)。据报道,肠道菌群的组成与ASD有关。肠道微生物群具有非常多样的组成,由细菌以及真菌,病毒和原生物组成(Enaud等,2018)。它与中枢神经系统有双向联系。例如,最近的文献计量学肠道中的数百万个神经细胞形成肠神经系统,该系统被认为是第二个大脑(Gershon,1999)。已经研究了微生物群 - 脑轴,该途径的双向通信是通过各种机制发生的,包括肠神经系统,自主神经系统,免疫系统,免疫系统,激素和神经递质(Cryan和Dinan,2012年)。可能参与微生物元素参与ASD的发病机理,于1998年首次报道,当时Bolte(1998)提出了以下假设:TETANI神经毒素从胃肠道传递到中枢神经系统,通过迷走神经,引起ASD症状。在动物模型中已经研究了肠道微生物群和ASD之间的联系。一项在2019年发表的研究,该研究将人ASD患者从人ASD患者移植到无菌小鼠中揭示了受体动物中Hallmark自闭症行为的发展(Sharon等,2019)。 在人类研究中也报道了肠道菌群与ASD之间的关联。 例如,一项焦磷酸测序研究观察到,在ASD患者中,杆菌存在很高,而在健康对照组中,Firmicutes更为丰富(Finegold等,2010)。 鉴于与ASD和肠道微生物群有关的兴趣趋势的上升,值得在该研究领域的文献中确定最有影感的科学成就。 它可以可视化详细的结果,并帮助研究人员对领域中的研究轨迹有透彻的了解,并确定研究热点和差距。一项在2019年发表的研究,该研究将人ASD患者从人ASD患者移植到无菌小鼠中揭示了受体动物中Hallmark自闭症行为的发展(Sharon等,2019)。在人类研究中也报道了肠道菌群与ASD之间的关联。例如,一项焦磷酸测序研究观察到,在ASD患者中,杆菌存在很高,而在健康对照组中,Firmicutes更为丰富(Finegold等,2010)。鉴于与ASD和肠道微生物群有关的兴趣趋势的上升,值得在该研究领域的文献中确定最有影感的科学成就。它可以可视化详细的结果,并帮助研究人员对领域中的研究轨迹有透彻的了解,并确定研究热点和差距。文献计量分析是一种广泛使用的严格方法,用于探索广泛的科学数据集并提取有用的信息,例如作者名称,全部引用和国家分布(Donthu等,2021)。