XiaoMi-AI文件搜索系统
World File Search SystemRANKL
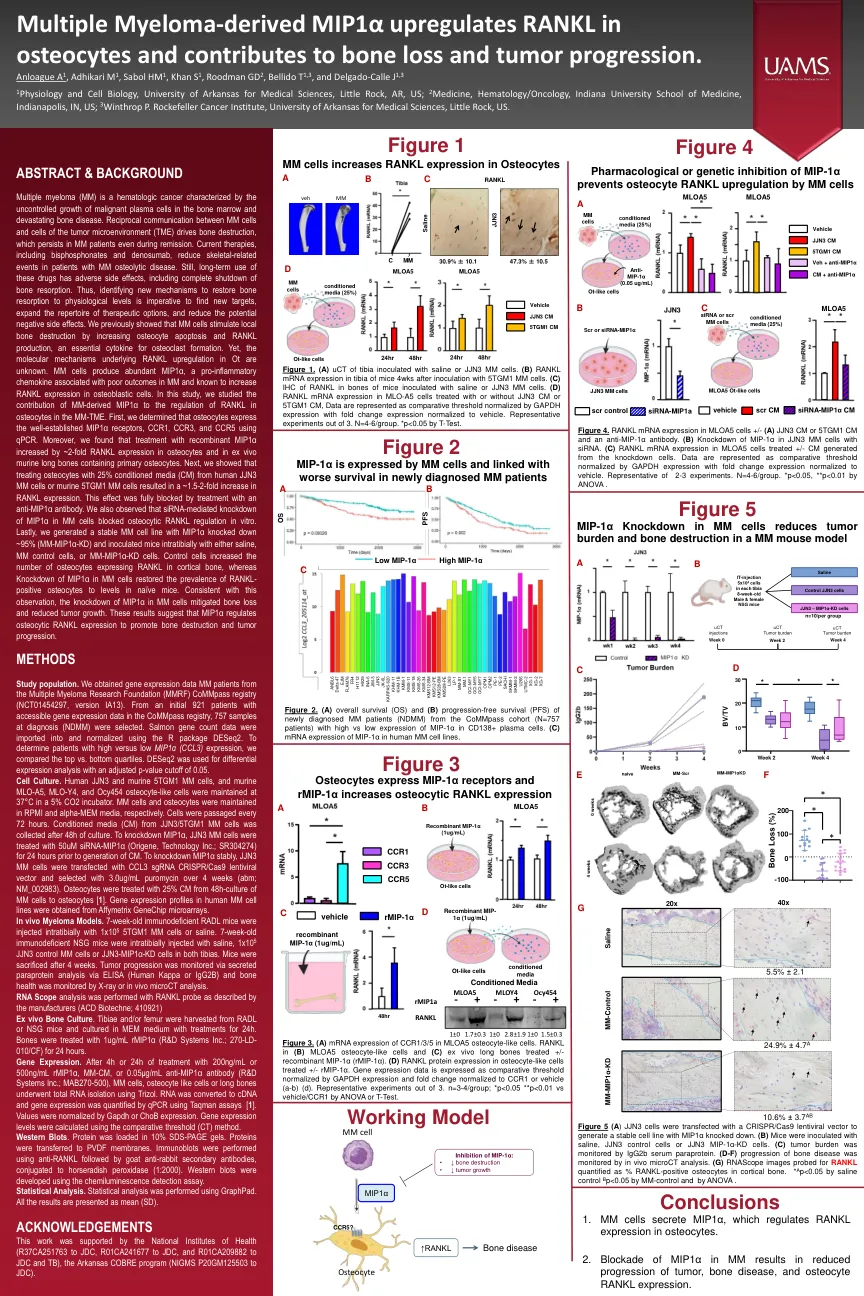
多发性骨髓瘤的MIP1α上调RANKL ...
多发性骨髓瘤(MM)是一种血液学癌,其特征是骨髓和毁灭性骨病中恶性血浆细胞的生长不受控制。MM细胞与肿瘤微环境(TME)细胞之间的相互通信驱动骨骼破坏,即使在缓解过程中,MM患者也持续存在。当前的疗法,包括双膦酸盐和denosumab,减少了MM骨化疾病患者的骨骼相关事件。仍然,这些药物的长期使用具有不利的副作用,包括骨吸收的完全关闭。因此,必须确定新的机制以将骨吸收恢复到生理水平,这对于寻找新靶标,扩展治疗选择的曲目并减少潜在的负面副作用至关重要。我们先前表明,MM细胞通过增加骨细胞凋亡和RANKL产生来刺激局部骨破坏,这是破骨细胞形成的必不可少的细胞因子。然而,OT中RANKL上调的分子机制尚不清楚。mM细胞产生丰富的MIP1α,这是一种与MM结局不佳相关的促炎性趋化因子,并且已知会增加成骨细胞中RANKL表达。在这项研究中,我们研究了MM衍生的MIP1α对MM-TME中骨细胞中RANKL调节的贡献。首先,我们确定骨细胞使用qPCR表达了良好的MIP1α受体CCR1,CCR3和CCR5。此外,我们发现重组MIP1α的治疗在骨细胞和含有原发性骨细胞的离体鼠长骨中增加了约2倍的RANKL表达。接下来,我们表明,从人JJN3 MM细胞或鼠5TGM1 MM细胞中使用25%条件培养基(CM)处理骨细胞,RANKL表达增加了约1.5-2倍。用抗MIP1α抗体处理完全阻断了这种作用。我们还观察到siRNA介导的MM细胞中MIP1α的敲低阻断了体外骨细胞RANKL调节。最后,我们生成了一个稳定的MM细胞系,MIP1α击倒了〜95%(MM-MIP1α-KD),并用盐水,MM对照细胞或MM-MIP1α-KD细胞在室内接种小鼠。对照细胞增加了在皮质骨中表达RANKL的骨细胞的数量,而MM细胞中MIP1α的敲低使RANKL-阳性骨细胞的患病率恢复为幼稚小鼠的水平。与该观察结果一致,MM细胞中MIP1α的敲低减轻骨质流失和肿瘤生长减少。这些结果表明MIP1α调节骨细胞RANKL表达以促进骨骼破坏和肿瘤进展。

基于单细胞打印技术和乳液偶联分析对 CRISPR/Cas9 RANKL 敲除间充质干细胞克隆进行表征,作为单细胞克隆的低细胞工作流程
基因改造单细胞的同质性对于许多应用(例如细胞系开发、基因治疗和组织工程,尤其是再生医学应用)而言是必需的。缺乏有效分离和表征 CRISPR/Cas9 工程细胞的工具被认为是这些应用中的一个重大瓶颈。尤其是蛋白质检测技术不兼容,无法在没有先决条件大规模克隆扩增的情况下确认蛋白质表达变化,这在许多应用中造成了僵局。为了改善工程细胞的表征,我们提出了一种改进的工作流程,包括基于高产量荧光特性的单细胞打印/分离技术、基因组编辑筛选(测量测定)、评估改变的基因表达的 mRNA rtPCR 和一种称为乳化偶联的多功能蛋白质检测工具,以提供高含量、统一的单细胞工作流程。该工作流程以 RANKL 敲除永生化间充质干细胞的工程和功能验证为例,这些细胞的骨形成能力发生了改变。由此产生的工作流程经济实惠,无需大规模克隆扩增细胞,整体克隆效率高于 CRISPR/Cas9 编辑细胞的 30%。尽管如此,由于单细胞克隆在细胞发育的早期高度并行阶段得到全面表征,包括 DNA、RNA 和蛋白质水平,因此该工作流程可提供更多成功编辑的细胞以供进一步表征,从而降低开发过程中后期失败的可能性。

简历教授Josef Penninger博士
Josef Penninger操纵小鼠基因,并研究这些突变对生物体发展和疾病出现的影响。主要重点是心脏和肺部疾病,癌症和骨骼疾病。他发现了负责骨变性的RANKL蛋白(骨质疏松症)。在健康的骨骼中,建立骨骼的细胞与细胞分解的细胞之间存在平衡。RANKL将血干细胞转化为吞噬细胞。 基于此,开发了一种阻断RANKL蛋白的药物,从而缓解了骨质疏松症。RANKL将血干细胞转化为吞噬细胞。基于此,开发了一种阻断RANKL蛋白的药物,从而缓解了骨质疏松症。基于此,开发了一种阻断RANKL蛋白的药物,从而缓解了骨质疏松症。

疫苗不平等和犹豫持续存在 - 我们必须解决这两个
最近的研究表明RANKL/RANK信号通路与能量代谢之间存在关联。在一项基于人群的大型研究中,较高的RANKL水平与在五年的随访期内增加了2型糖尿病风险的四倍。6 7 RANKL信号的下调可以改善小鼠和人类的葡萄糖代谢。7-9在肝脏中抑制了RANKL信号传导,肝胰岛素敏感性和血浆葡萄糖浓度得到改善。7用denosumab封闭RANKL信号传导可以显着降低循环的二肽基肽酶4并增加胰高血糖素样肽-1(GLP-1)水平。9虽然在患有糖尿病的人群中尚未进行随机对照试验,但观察性研究的结果表明,在2型糖尿病或糖尿病前期的参与者中,葡萄糖稳态改善了,与9个月相比,在12个月中,与denosumab的参与者相比,在12个月中,与denosumab

良性骨肿瘤中denosumab的当前适应症
deosumab是一种完全人性化的单克隆抗体(核因子kappa b配体),可抑制等级 - rankl相互作用。骨(GCTB)过表达的巨细胞肿瘤的单核基质细胞,这是募集,形成,增强功能增强和骨细胞状巨细胞存活的必不可少的介体。为此,RANKL与巨细胞表面的等级相互作用。这种相互作用是由Denosumab与RANKL结合的。因此,破骨细胞从肿瘤组织中消失,并且主要被松散的结缔组织和新形成的骨骼所取代。H3F3A-阳性基质细胞,从而强调了denosumab在肿瘤基质细胞上的无效性(1)。

比较有或患有2型糖尿病的牙周炎患者的血清和牙龈5型牙龈液的炎症态
抽象背景/目的:牙周炎与2型糖尿病之间存在双向关系。这项研究旨在比较患有或患有2型糖尿病(T2DM)和健康受试者的牙周炎患者的血清和牙龈循环疗法(GCF)中的炎症状态。材料和方法:20名受试者是系统的,牙周健康(H组),40名受试者患有牙周炎(CP组),其他40名患有牙周炎和2型糖尿病(DC组)。禁食血糖(FBG)和HBA1C。测量了 GCF和白介素(IL)17的血清水平,Visfatin,核因子-Kappa B(NF-K B)配体(RANKL)/骨蛋白酶蛋白蛋白蛋白蛋白蛋白酶(OPG)的受体激活剂(NF-K B)。结果:GCF的总量,IL-17的总量,GATSATIN,RASTATIN/OPG比例的GCF及其在CP和DC组中的血清浓度高(P <0.05),而H组的DC组也高于CP组的GCF组(p <0.05)(p <0.05)。在PD 3 mM,GCF体积,IL-17,Visfatin和RANKFATIN和RANKL/OPG比的样本位点上,DC组和CP组的比率高(P <0.05)(p <0.05)高于H组,在DC

TGF-B激活的激酶-1抑制剂LL-Z1640-2降低关节
目标。异常NLRP3炎症症状激活,这可能有助于使炎症和骨骼破坏衰弱。在这里,我们探讨了有效的TGF-B激活激活的激活激酶-1(TAK1)抑制剂LL-Z1640-2(LLZ)对胶原蛋白诱导的关节炎(CIA)的关节膨胀和骨破坏的效率。方法。ll-Z1640-2每隔一天在中央情报局小鼠中一次施用。进行了临床和组织学评估。启动和激活NLRP3炎症和骨质质构成活性。结果。nlrp3炎症形成。TACE和RANKL在CIA关节中分别在滑膜巨噬细胞和纤维细胞中过表达。使用LLZ治疗可缓解上述所有变化。结果,LLZ明显抑制了滑膜肥大和pannus形成,以减轻CIA小鼠的疼痛和炎症。llz可以阻止RAW264.7巨噬细胞系,原代骨髓巨噬细胞和LPS治疗后的NLRP3炎症的启动和激活,从而抑制其IL-1 B产生。llz还抑制了LPS诱导的骨髓巨噬细胞中TACE和TNF-a的产生,并废除了IL-1 B-诱导的MMP-3,IL-6和RANKL的产生。此外,LLZ直接抑制RANKL介导的OC形成和激活。结论。tak1抑制LLZtak1抑制LLZ

骨头
脑衍生的神经营养因子(BDNF)是一种神经营养蛋白,在中枢神经系统和周围组织中表达,受到GSα /CAMP途径的调节。在骨骼中,它调节成骨,并刺激骨化肿瘤(如多发性骨髓瘤)中的RANKL分泌和破骨细胞形成。纤维发育不良(FD)是由GSα基因的功能收益突变引起的罕见的骨骼遗传疾病,其中RANKL依赖性增强的骨吸收是骨骼脆弱性和临床发病率的主要原因。我们观察到BDNF转录本在人类FD病变中表达。具体而言,对从FD患者获得的活检进行的免疫定位研究揭示了成骨细胞中BDNF的表达,并且在纤维组织内的纺锤形细胞中的表现较低。因此,我们假设BDNF可以通过刺激RANKL分泌和骨吸收来在FD的发病机理中发挥作用。为了测试这种疗法,我们使用了人类疾病的EF1α-GSαR201C小鼠模型(FD小鼠)。Western印迹分析显示,与WT小鼠相比,FD小鼠的骨段中BDNF的表达更高,而小鼠FD病变中的免疫标记模式与在人FD中观察到的相似。用抗BDNF的单克隆抗体对FD小鼠进行处理,可减少纤维组织,以及股骨病变内的破骨细胞和骨爆炸的数量。这些结果揭示了BDNF是FD发病机理的新玩家,并且可以在FD骨骼病变中滋养破骨细胞生成的潜在分子机制。他们还建议BDNF抑制作用可能是减少FD中异常骨骼重塑的一种新方法。

denosumab的处方框架(Prolia
deosumab是一种完全人类的单克隆抗体,通过中和RANKL抑制骨吸收,RANKL是破骨细胞形成,功能和存活的关键介体。Demumab获得男性和女性的骨质疏松症治疗。NICE TA204涵盖了绝经后妇女的原发性和继发性骨质疏松骨折,其骨折风险增加。本文档旨在为GPS开处方Denosumab提供一个框架。它阐明了进入共享护理安排的全科医生和医院专家的相关责任。该文件应与EL(91)127中给出的规定事项的一般指南一起阅读,“医院和GPS之间的处方责任”。交通信号灯分类请注意,此共享护理框架是指denosumab 60mg注射(Prolia),该注射(Prolia)被批准为Hull和East Riding中的琥珀色药物。Demumab 120mg注射(XGEVA)被许可用于减少实体瘤骨转移患者的骨骼损伤,被归类为红色药物,不应由非专家团队开处方。