XiaoMi-AI文件搜索系统
World File Search SystemRNase
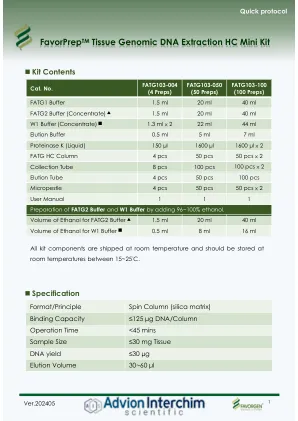
forprepreptm组织基因组DNA提取HC Mini Kit
6。新制备FATG1-PK混合物,预先330μLFATG1缓冲液,30 µL蛋白酶K和8 µL 50 mg/ml RNase A(如果需要无RNA的基因组DNA,则需要每个样品),然后执行DNA提取之前,每个样品需要无RNA的基因组DNA)。
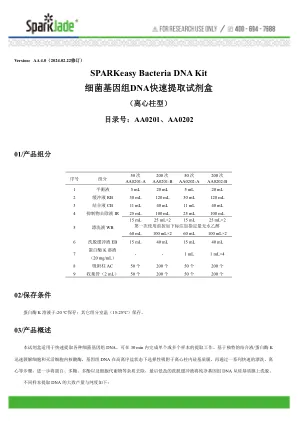
SPARKeasy Bacteria DNA Kit 细菌基因组DNA快速 ...
▲ 可选步骤:如果 RNA 残留较多,需要去除 RNA ,可以在加入 200 μL 结合液 CB 前加 4 μL RNase A ( 100 mg/mL )溶液,振荡混匀,室温放置 5-10 min 。
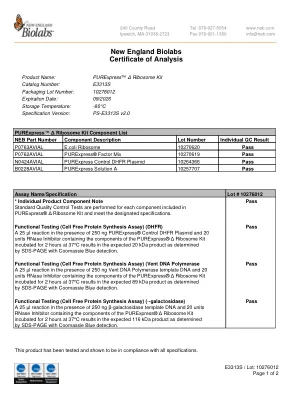
新英格兰Biolabs分析证书
A 25 µL反应和20个单位RNase抑制剂,其中含有Purexpress®Δ核糖体的成分在37°C下在37°C下孵育2小时,从而在预期的20 kDa产物中通过SDS-PAGE与Coomassie Blue blue detection确定,导致预期的20 kDa产物。
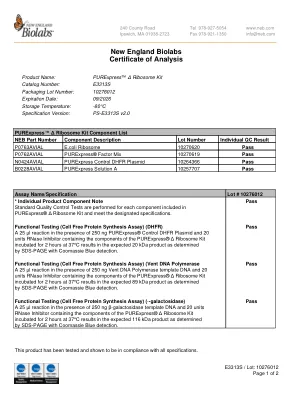
新英格兰生物实验室分析证书
在 250 ng PURExpress® 对照 DHFR 质粒和 20 单位 RNase 抑制剂(含有 PURExpress® Δ 核糖体试剂盒的成分)存在下进行 25 µl 反应,在 37°C 下孵育 2 小时,通过 SDS-PAGE 和考马斯亮蓝检测测定,可得到预期的 20 kDa 产物。

牛骨骼肌基因组DNA
牛骨骼肌基因组DNA是一种高度完整的高分子大小DNA。它是从单个供体的单个组织中提取的,并用无DNase RNase处理以去除污染物RNA。基因组DNA精确地通过纳米体(一种分光光度计技术)测量,并存储在-80oC中。
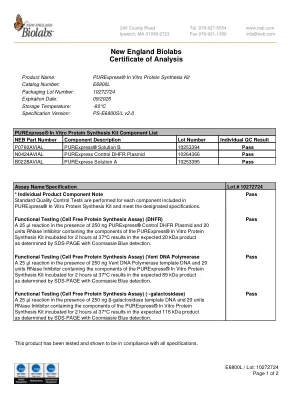
新英格兰生物实验室分析证书
在 250 ng PURExpress® 对照 DHFR 质粒和 20 单位 RNase 抑制剂(包含 PURExpress® 体外蛋白质合成试剂盒的成分)存在下进行 25 µl 反应,在 37°C 下孵育 2 小时,通过 SDS-PAGE 和考马斯亮蓝检测确定可得到预期的 20 kDa 产物。

我们如何实施新疗法来改变这种模式?
Ago2,argonaute 2;又名;ASO,反义寡核苷酸;mRNA,信使 RNA;RISC,RNA 诱导沉默复合物;RNase H1,核糖核酸酶 H1;siRNA,小干扰 RNA。图片改编自:Ginsburg 等人 (2017),基因组和精准医学基础,翻译和实施。爱思唯尔

新英格兰生物实验室分析证书
在 250 ng vTPA(截短组织型纤溶酶原激活剂)模板质粒 DNA、1 µl 增强剂 1、1 µl 增强剂 2 和 20 单位 RNase 抑制剂(含有 PURExpress® 体外蛋白质合成试剂盒的成分)存在下进行 25 µl 反应,在 37°C 下孵育 2 小时,通过 SDS-PAGE 和考马斯亮蓝检测测定,可得到预期的 40 kDa 产物。


海洋浮游植物中的RNA和DNA测量的新双染色技术
RNA:DNA比率已被用作各种海洋器官中生长速率的生化指标(Sutcliffe 1970,Buckley&Lough 1987,Bulow 1987,Clemmesen 1987,Clemmesen 1987,1988,Clarke等,Clarke等,1988,Raae等。 1988,Mordy&Carlson 1991),包括Phyto-Plankton(Dortch等人 1983,1985,Mordy&Carlson 1991)。 由于RNA和DNA的化学相似性,通过经典方法分别量化每种都需要冗长的提取。 此外,通过分光光度法(紫外线浸泡)和量热法(使用RNA和二苯胺的脑甲醇用于DNA)的最终量化很困难,从而使低剂量和干扰因提取其他细胞成分以及除核酸以外的许多细胞成分(例如 Herbert等。 1971,Cattolico 1978)。 可以通过使用荧光染色来实现更大的敏感性,但是可用的方法仍然是某种程度上 - 繁琐,耗时且遇到一些潜在问题(Holm-Hansen等人。 1968,Thoresen等。 1983,Clemmesen 1988)。 例如,在Prasad等人开发的广泛使用的方法中。 (1972),总核酸用溴化乙锭测量,溴化乙锭与DNA和RNA反应,并产生高度荧光化合物。 然后用酶RNase破坏RNA,并测量DNA引起的荧光。 然后从差异计算 RNA con-中心1988,Raae等。1988,Mordy&Carlson 1991),包括Phyto-Plankton(Dortch等人 1983,1985,Mordy&Carlson 1991)。 由于RNA和DNA的化学相似性,通过经典方法分别量化每种都需要冗长的提取。 此外,通过分光光度法(紫外线浸泡)和量热法(使用RNA和二苯胺的脑甲醇用于DNA)的最终量化很困难,从而使低剂量和干扰因提取其他细胞成分以及除核酸以外的许多细胞成分(例如 Herbert等。 1971,Cattolico 1978)。 可以通过使用荧光染色来实现更大的敏感性,但是可用的方法仍然是某种程度上 - 繁琐,耗时且遇到一些潜在问题(Holm-Hansen等人。 1968,Thoresen等。 1983,Clemmesen 1988)。 例如,在Prasad等人开发的广泛使用的方法中。 (1972),总核酸用溴化乙锭测量,溴化乙锭与DNA和RNA反应,并产生高度荧光化合物。 然后用酶RNase破坏RNA,并测量DNA引起的荧光。 然后从差异计算 RNA con-中心1988,Mordy&Carlson 1991),包括Phyto-Plankton(Dortch等人1983,1985,Mordy&Carlson 1991)。 由于RNA和DNA的化学相似性,通过经典方法分别量化每种都需要冗长的提取。 此外,通过分光光度法(紫外线浸泡)和量热法(使用RNA和二苯胺的脑甲醇用于DNA)的最终量化很困难,从而使低剂量和干扰因提取其他细胞成分以及除核酸以外的许多细胞成分(例如 Herbert等。 1971,Cattolico 1978)。 可以通过使用荧光染色来实现更大的敏感性,但是可用的方法仍然是某种程度上 - 繁琐,耗时且遇到一些潜在问题(Holm-Hansen等人。 1968,Thoresen等。 1983,Clemmesen 1988)。 例如,在Prasad等人开发的广泛使用的方法中。 (1972),总核酸用溴化乙锭测量,溴化乙锭与DNA和RNA反应,并产生高度荧光化合物。 然后用酶RNase破坏RNA,并测量DNA引起的荧光。 然后从差异计算 RNA con-中心1983,1985,Mordy&Carlson 1991)。由于RNA和DNA的化学相似性,通过经典方法分别量化每种都需要冗长的提取。此外,通过分光光度法(紫外线浸泡)和量热法(使用RNA和二苯胺的脑甲醇用于DNA)的最终量化很困难,从而使低剂量和干扰因提取其他细胞成分以及除核酸以外的许多细胞成分(例如Herbert等。 1971,Cattolico 1978)。 可以通过使用荧光染色来实现更大的敏感性,但是可用的方法仍然是某种程度上 - 繁琐,耗时且遇到一些潜在问题(Holm-Hansen等人。 1968,Thoresen等。 1983,Clemmesen 1988)。 例如,在Prasad等人开发的广泛使用的方法中。 (1972),总核酸用溴化乙锭测量,溴化乙锭与DNA和RNA反应,并产生高度荧光化合物。 然后用酶RNase破坏RNA,并测量DNA引起的荧光。 然后从差异计算 RNA con-中心Herbert等。1971,Cattolico 1978)。 可以通过使用荧光染色来实现更大的敏感性,但是可用的方法仍然是某种程度上 - 繁琐,耗时且遇到一些潜在问题(Holm-Hansen等人。 1968,Thoresen等。 1983,Clemmesen 1988)。 例如,在Prasad等人开发的广泛使用的方法中。 (1972),总核酸用溴化乙锭测量,溴化乙锭与DNA和RNA反应,并产生高度荧光化合物。 然后用酶RNase破坏RNA,并测量DNA引起的荧光。 然后从差异计算 RNA con-中心1971,Cattolico 1978)。可以通过使用荧光染色来实现更大的敏感性,但是可用的方法仍然是某种程度上 - 繁琐,耗时且遇到一些潜在问题(Holm-Hansen等人。1968,Thoresen等。 1983,Clemmesen 1988)。 例如,在Prasad等人开发的广泛使用的方法中。 (1972),总核酸用溴化乙锭测量,溴化乙锭与DNA和RNA反应,并产生高度荧光化合物。 然后用酶RNase破坏RNA,并测量DNA引起的荧光。 然后从差异计算 RNA con-中心1968,Thoresen等。1983,Clemmesen 1988)。 例如,在Prasad等人开发的广泛使用的方法中。 (1972),总核酸用溴化乙锭测量,溴化乙锭与DNA和RNA反应,并产生高度荧光化合物。 然后用酶RNase破坏RNA,并测量DNA引起的荧光。 然后从差异1983,Clemmesen 1988)。例如,在Prasad等人开发的广泛使用的方法中。(1972),总核酸用溴化乙锭测量,溴化乙锭与DNA和RNA反应,并产生高度荧光化合物。然后用酶RNase破坏RNA,并测量DNA引起的荧光。RNA con-中心