XiaoMi-AI文件搜索系统
World File Search SystemSMC3

解析唐氏综合征相关白血病模型中巨核细胞生成的逐步突变损伤
突变并可以检查巨核细胞分化和其他疾病表型的渐进性扰动。在本期的 JCI 中,Arkoun 和同事使用分步技术将 GATA1 、 MPL 和 SMC3 突变体引入患有或不患有 DS 的人的诱导多能干细胞 (iPSC) 中实现了这一目标 (9)。研究人员揭示了每种变体的个体贡献以及它们如何与 T21 协同导致 DS-AMKL。作者使用 CRISPR/Cas9 技术进行分步基因编辑,生成了 20 个不同的二体和三体 iPSC 克隆,这些克隆包含 GATA1、MPL W515K 和 SMC3 杂合缺失 (SMC3 +/–) 的组合,并通过功能分析验证了这些变化。 MPL 是血小板生成素的跨膜受体,是巨核细胞成熟为血小板所必需的。胞内结构域通过与 JAK2 相互作用介导信号传导。MPL 515 位点的多个功能获得性氨基酸置换通过血小板生成素依赖性激活 JAK/STAT 通路导致骨髓增生性疾病 (10)。有趣的是,W515K/L 突变也见于 T21 患者的 AMKL 和获得额外 21 号染色体的整倍体个体 (D21) 的白血病中,这可能导致巨核细胞分化改变 (7, 11)。T21 和 Gata1 背景下的 MPL 突变足以诱发小鼠巨核细胞白血病 (12)。此外,作者假设,黏连蛋白基因 SMC3 的单倍体不足通过杂合失活会改变 GATA1 结合的染色质可及性,从而改变巨核细胞分化的转录控制。鉴于这些突变单独导致髓系谱系破坏,逐步 iPSC 模型

从结构特征到生物学功能
1.大多数回路是短的(<2 Mb),并且在人类和小鼠之间,在细胞类型之间得到强烈保守。2.锚定在启动子上的环与增强子和基因激活增加有关。3。环路经常划分接触域的边界4.CTCF和粘蛋白亚基RAD21和SMC3与环相关;这些蛋白质中的每一个都以超过86%的环锚固量发现。
分析提供获得的疾病(血液学)
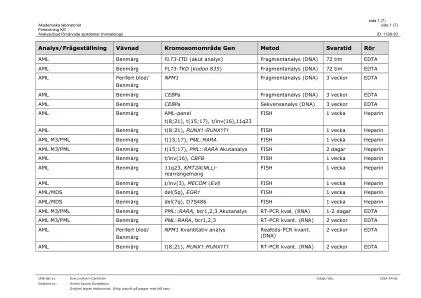
leukemiutringbenmärg/blod mylood面板*(ABL1,ANKRD26,ASXL1,ATRX,BCOR,BCOR,BCORL1,BRAF,CALR,CALR,CBL,CBL,CBL,CBL,CDKN2A,CDKN2A,CEBPA,CEBPA,CEBPA,CSF3R,CSF3R,CSF3R,CUX1,DDX41,DDX41,DNMT3A fbxw7, FLT3, GATA1, GATA2, GNAS, HRAS, Idh1, Idh2, Ikzf1, jak2, jak3, kdm6a, kit, kraas, kmt2a, mpl, myd88, NF1, Notch1 (INKLUSIVE 3´UTR), NPM1, NRAS, PDGFRA, PHF6, PPM1D, Pten, Ptpn11, Rad21, Runx1, Samd9, SAMDL9, Setbp1, SF3B1, SMC1A, SMC3, SRSF2, Stag2, Stat3, Stat5B, Tet2, TP53, U2AF1, WT1, ZRSR2, BTK, plcg2, terc) Div>

2021 年 ASPHO 会议论文和海报索引
全体会议论文 # 2001 黏连蛋白功能改变对核心结合因子急性髓系白血病增殖的影响 Shannon Conneely、Jason Rogers、Matthew Miller、Jason Guo、Rohit Gupta、Geraldo Medrano、Debananda Pati、Rachel Rau 贝勒医学院/德克萨斯儿童医院,美国德克萨斯州休斯顿 背景:核心结合因子急性髓系白血病 (AML) 是一种常见的儿童 AML,其特征是 inv(16) 或 t(8;21) 病变,这些病变会抑制核心结合因子复合物的功能。尽管这些重排被认为是 AML 的有利风险,但近 30% 的核心结合因子 AML 儿童会复发,这表明需要继续加深对 AML 生物学的了解和寻找新的治疗靶点。黏连蛋白复合体基因突变常见于 t(8;21) AML,但在 inv(16) AML 中从未发现,这表明黏连蛋白在每种核心结合因子 AML 亚型的病理生理学中发挥着独特的作用。目标:本项目的目标是确定黏连蛋白突变如何改变核心结合因子 AML 的生物学特性。我们假设,黏连蛋白正常功能的丧失会增强表达 t(8;21) AML 特征性 RUNX1-CBFA2T1 (RC) 融合蛋白的细胞增殖,并抑制表达 inv(16) AML 特征性 CBFß-SMMHC (CS) 融合的细胞的增殖能力。设计/方法:从黏连蛋白正常 (Smc3 +/+) 或黏连蛋白单倍体不足 (Smc3 +/-) 的小鼠体内采集骨髓细胞。我们利用逆转录病毒转导来表达空载体对照、RC 融合或 CS 融合蛋白。然后将转导的细胞接种在含有干细胞和骨髓促进细胞因子的甲基纤维素中,进行连续接种试验,或移植到致死性辐射受体小鼠体内,以评估对白血病转化的影响。结果:连续接种试验表明,黏连蛋白单倍体不足会增加表达 RC 蛋白的细胞的集落形成能力,并降低表达 CS 蛋白的细胞的集落形成能力。黏连蛋白单倍体不足会改变几种关键造血调节基因的表达,尽管这些影响取决于存在哪种融合蛋白。在小鼠 RC 模型中,无论黏连蛋白功能如何,都会发展为未分化白血病。然而,二次移植模型显示,黏连蛋白功能下降会导致白血病存活时间缩短,骨髓浸润增加。结论:正常黏连蛋白功能的丧失对表达核心结合因子 AML 融合蛋白的细胞增殖有不同的影响。在表达与 t(8;21) AML 相关的 RC 融合的细胞中,黏连蛋白功能的降低在白血病转化之前提供了生长优势,并带来了更具浸润性和侵袭性的白血病表型。或者,黏连蛋白功能下降导致表达 inv(16) AML CS 融合的细胞生长不利,造血基因表达发生显著变化。未来的实验将重点阐明核心结合因子 AML 中黏连蛋白功能下降所改变的潜在细胞机制。

粘合素超元在循环挤出过程中DNA
抽象的粘着蛋白将基因组DNA挤压成促进染色质组装,基因调节和重组的环。在这里,我们表明粘着蛋白将负超胶引入挤出的DNA中。超螺旋需要粘蛋白的ATPase头,这些头部夹紧DNA以及在粘蛋白的铰链上的DNA结合位点,表明在铰链和夹具之间约束粘蛋白超侧Coil DNA。我们的结果表明,一旦粘蛋白在超涂层期间达到其失速扭矩,DNA挤出会停止,而粘蛋白突变体预测会停滞在较低的扭矩形成细胞中的较短环。这些结果表明,超涂层是环挤出机制的组成部分,并且粘着蛋白不仅通过循环DNA,而且通过将其超级旋转来控制基因组结构。真核间相细胞中的主要文本,SMC(“染色体的结构维持”)复合粘着蛋白将基因组DNA折叠成环和拓扑结构域(TADS;参考(1-4)),可以调节转录(5),重组(6,7),姐妹染色单体分离(8)和复制(9)。粘着蛋白通过由ATP结合 - 水溶液周期控制的构象变化(12)(在(13)中进行了综述),将DNA挤压为环(10,11)。这些是由粘蛋白的SMC1和SMC3亚基催化的,其中包含50 nm长的盘绕螺旋,二聚体“铰链”结构域和球形ATPase'heads'(图s1a),与ABC转运蛋白相关(14)。在ATP结合后,粘蛋白的头部接合和一个称为NIPBL“夹具” DNA的亚基在接合的ATPase头顶上(参考(12,15-17);如图。s1b)。这些动作产生〜15 pn力(18)和循环挤出步骤〜40 nm(100-200 bp;ref。(19)),表明在头部互动过程中将DNA卷入形成循环中。相比之下,在环挤出过程中DNA的构象变化知之甚少。拓扑异构酶II在粘着蛋白环的底部结合并切割DNA(20-23),这表明DNA在这些位点上是超螺旋的。有丝分裂SMC复合物冷凝蛋白还与拓扑异构酶(24-30)共定位并相互作用,并且可以在体外超涂DNA(31-33)。已经提出了此过程发生在循环挤出过程中(31,33),但发现粘着蛋白不适合