XiaoMi-AI文件搜索系统
World File Search SystemUSH2A
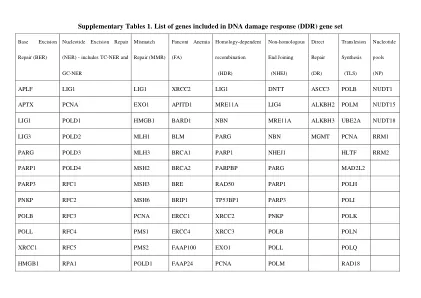
补充表 1. DNA 损伤反应 (DDR) 基因集中包含的基因列表
基因 p HR 基因 p HR ABCB11 0.00000 18.2 CREBBP 0.93 1.05 ABL1 0.00004 10.5 KIF21A 0.50 1.53 ARHGEF16 0.00000 14.2 KMT2E 0.65 1.32 C5orf42 0.00023 17.7 MKI67 0.04 3.18 CCDC88C 0.00028 8.65 OTOG 0.66 0.746 CEP350 0.00003 11.2 POLQ 0.57 1.48 CHD4 0.00029 10.6 SEMA5A 0.22 0.291 CREBBP 0.004 7.22 SON 0.10 3.91e−09 CTAGE1 0.00039 8.38 SPEF2 0.87 1.12 DHX9 0.00007 10.4 STAB2 0.75 0.822 DMD 0.00021 8.93 TCHH 0.03 3.18 DNAH6 0.00022 11 TPR 0.003 4.89 FAM124A 0.00028 8.65 UNC13C 0.94 1.05 FBN1 0.00078 7.62 USH2A 0.73 0.82 GREB1L 0.00025 10.9 ZNF236 0.17 2.2 KIAA1109 0.00237 1.68E+09 KIF21A 0.002 8.24 KMT2E 0.003 7.56 MKI67 0.003 7.66 NBAS 0.00001 15 NWD2 0.00068 7.93 OTOG 0.002 8.24 PARP14 0.00045 9.92 POLQ 0.00073 7.75 SDE2 0.00001 11.8 SEMA5A 0.00030 8.74 SH3TC2 0.00000 17.1

视网膜纤毛病和潜在的基因疗法
摘要:人类感光细胞的功能依赖于高度特化的纤毛。纤毛功能的紊乱通常会导致感光细胞死亡和视力丧失。视网膜纤毛病是一种遗传多样性的视网膜遗传病,会影响感光细胞纤毛的各个方面。尽管利用动物疾病模型对视网膜纤毛病的理解取得了进展,但它们往往无法准确模拟观察到的患者表型,这可能是由于结构和功能与人类视网膜存在偏差。人类诱导多能干细胞 (hiPSC) 可用于生成替代疾病模型,即 3D 视网膜类器官,其中包含所有主要的视网膜细胞类型,包括带有纤毛结构的感光细胞。这些视网膜类器官有助于研究人类衍生系统中的疾病机制和潜在疗法。三维视网膜类器官仍是一项发展中的技术,尽管取得了令人瞩目的进展,但仍存在一些局限性。本综述将讨论 hiPSC 衍生的视网膜类器官技术现状,该技术可准确模拟与基因(包括 RPGR 、 CEP290 、 MYO7A 和 USH2A )相关的主要视网膜纤毛病。此外,我们还将讨论针对视网膜纤毛病的新型基因治疗方法的开发,包括大基因的传递和基因编辑技术。

综合征和非...
目的:本研究旨在使用100,000个基因组项目(100kgp)的数据来描述英国人群中综合征和非综合性听力损失(HL)的遗传格局。设计:队列研究环境:NHS英格兰参与者:2013年至2018年之间,有2,271个患有综合症和非综合症HL的家庭。包括至少一个人类表型本体论(HPO)术语“听力障碍”(HP:0000365)的后代;这相当于5,488个人,其中包括2,762个受影响的个体和2,726个未受影响的亲戚。主要结局指标:通过整个基因组测序确定的听觉表型的诊断率和不同基因诊断的患病率。结果:总体诊断产率保守估计为27.5%(625/2271),在273种不同的基因中鉴定出诊断。常见的致病基因包括USH2A,GJB2,COL1A1和MYO15A,约占诊断的20%。此诊断率不包括不确定意义的变体(VUS),基因中HL不能自信地归因于已确定的变体或仍在等待确认的变体。包含这些类别将使诊断产量增加到39.6%。这项工作描述了100kGP标准管道和补充分析,包括使用Exomiser。分层允许定量具有特定表型组合的遗传诊断可能性,并鉴定通过听觉表型遗传诊断的阳性预测因子。报告了先天性(33.2%),双侧(27%)和高频(32.4%)听证子类型的人的诊断率显着提高。此外,在仅限于听觉系统的HPO术语患者中,大约40%的诊断归因于可能具有更广泛综合征表型的基因(非综合模仿)。在耳朵和眼睛异常的患者中出现高诊断产量(56%),这在很大程度上是由与Usher和Wolfram综合征相关的基因驱动的。结论是,这项研究为综合和非综合征HL的复杂基因组和表型结构提供了宝贵的见解,该结构具有改善诊断管道和临床护理的潜力。