XiaoMi-AI文件搜索系统
World File Search SystemUSP1

ASN-3186是...
24 24 24 24泛素特异性肽酶1(USP1)是DNA转移合成的关键调节剂和Fanconi贫血DNA Repition途径1,2。USP1从多种底物(PCNA,FANCI,FANCD2,PARP1,EZH2,CHK1等)中去除泛素与DNA损伤修复(DDR)3非常重要。USP1抑制剂可能会患有DDR脆弱性的某些癌症。ASN-3186是去泛素化酶USP1的选择性和有效抑制剂。ASN-3186治疗导致BRCA1/2突变的乳腺癌细胞系中的细胞死亡。ASN-3186与第一代或第二代PARP抑制剂(Olaparib/saruparib)结合使用时表现出强大的细胞杀伤协同作用。此外,ASN-3186在BRCA1/2MUT和HRD-(同源重组缺乏症)中表现出强烈的肿瘤生长抑制作用,具有主要PARPI耐药性。在头对头研究中,ASN-3186被发现比KSQ-4279(据报道的USP1抑制剂)4作为单一疗法或与Brcamut肿瘤模型中的Olaparib结合使用。正在计划进一步开发ASN-3186作为潜在的一流USP1抑制剂。
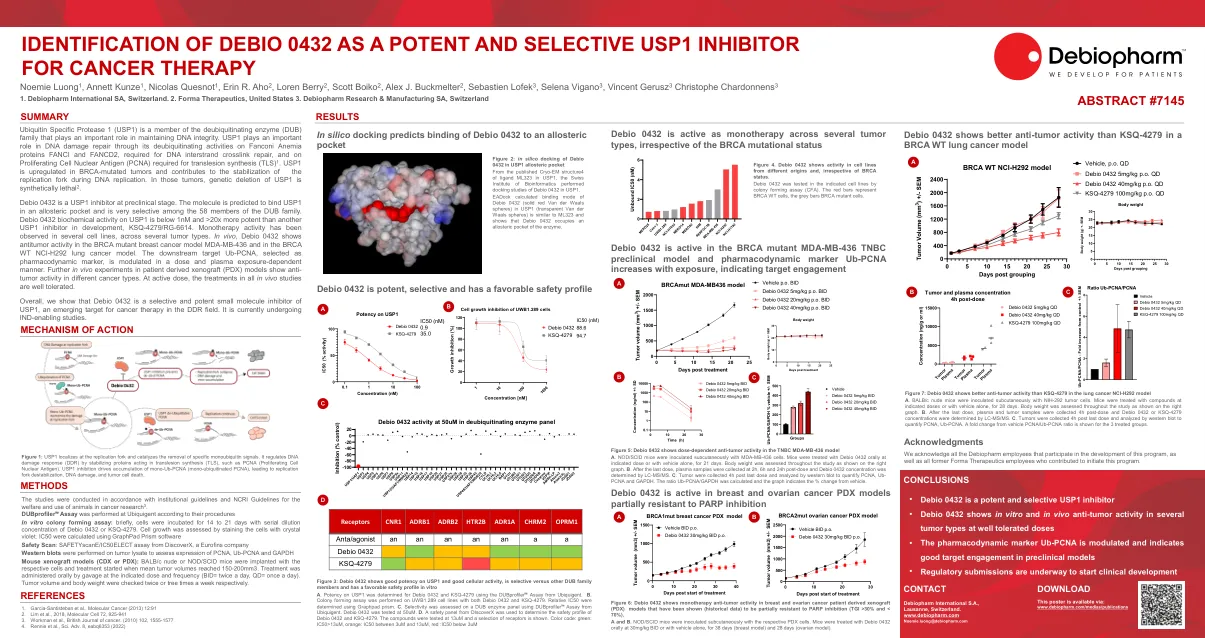
将Debio 0432识别为有效且有选择性的USP1 ...
Debio 0432是临床前阶段的USP1抑制剂。该分子预计将在变构袋中结合USP1,并且在DUB家族的58个成员中非常有选择性。DEBIO 0432 USP1上的生化活性低于1NM低于1NM,比其他USP1抑制剂(KSQ-4279/RG-6614)高20倍。单一疗法活性。在体内,Debio 0432显示了BRCA突变乳腺癌模型MDA-MB-436和BRCA WT NCI-H292肺癌模型中的抗肿瘤活性。以剂量和血浆暴露依赖性方式调节下游目标UB-PCNA,被选为药效标记。进一步的患者衍生异种移植物(PDX)模型的体内实验显示了不同癌症类型的抗肿瘤活性。在活性剂量时,所有体内研究中的治疗方法都得到很好的耐受性。

对临床USP1抑制剂作用机理的结构和生化见解,KSQ-4279
摘要:DNA损伤会触发介导修复的细胞信号传导级联。此信号在癌症中经常失调。介导该信号传导的蛋白质是治疗干预的潜在靶标。泛素特异性蛋白酶1(USP1)就是这样一个靶标,在临床试验中已经有小分子抑制剂。在这里,我们使用生化分析和冷冻电子显微镜(冷冻)来研究临床USP1抑制剂KSQ-4279(RO7623066),并将其与已建立的良好工具化合物进行比较。我们发现KSQ-4279与ML323的USP1同一隐性位点结合,但以微妙的方式破坏蛋白质结构。抑制剂结合驱动了USP1的热稳定性的大幅提高,USP1的热稳定性可以通过填充USP1中疏水隧道样口袋的抑制剂介导。我们的结果有助于理解分子水平USP1抑制剂的作用机理。■简介
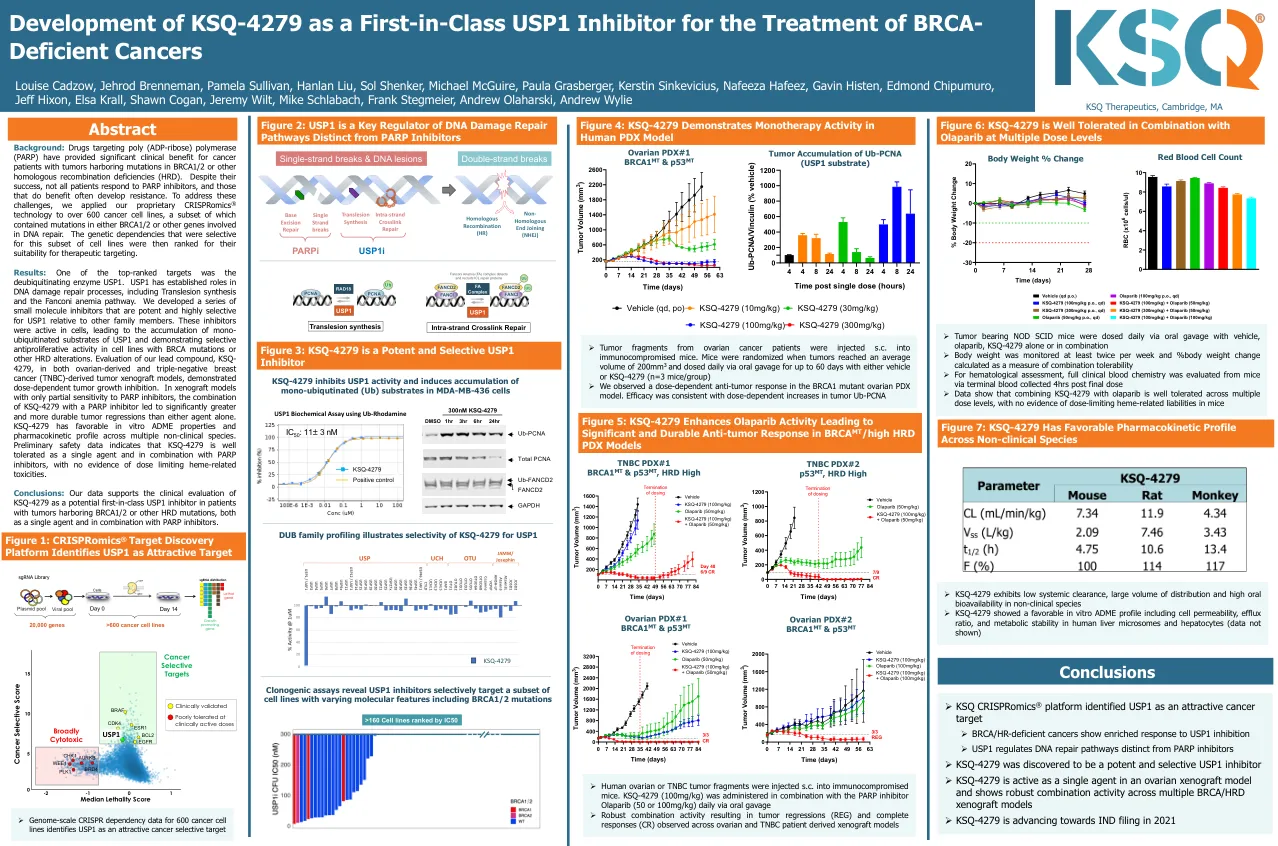
KSQ-4279 的开发作为治疗 BRCA 缺陷型癌症的首创 USP1 抑制剂
结果:排名靠前的靶标之一是去泛素化酶 USP1。USP1 在 DNA 损伤修复过程中发挥着重要作用,包括跨损伤合成和范康尼贫血途径。我们开发了一系列小分子抑制剂,这些抑制剂对 USP1 具有强效且高度选择性,相对于其他家族成员而言。这些抑制剂在细胞中具有活性,导致 USP1 单泛素化底物的积累,并在具有 BRCA 突变或其他 HRD 变异的细胞系中表现出选择性抗增殖活性。在卵巢衍生和三阴性乳腺癌 (TNBC) 衍生肿瘤异种移植模型中对我们的先导化合物 KSQ-4279 的评估表明,其具有剂量依赖性肿瘤生长抑制作用。在对 PARP 抑制剂仅部分敏感的异种移植模型中,KSQ-4279 与 PARP 抑制剂的组合比单独使用任何一种药物可显著改善肿瘤消退并延长其持续时间。 KSQ-4279 在多种非临床物种中具有良好的体外 ADME 特性和药代动力学特征。初步安全数据表明,KSQ-4279 作为单一药物和与 PARP 抑制剂联合使用具有良好的耐受性,没有证据表明存在剂量限制性血红素相关毒性。

去泛素化酶USP1在结直肠癌细胞中被ML323自动泛素化和不稳定
目标:我们先前的研究表明,USP1抑制剂ML323在结直肠癌(CRC)细胞中下调了USP1,但特定机制仍然未知。方法:将CRC细胞裂解以进行免疫印迹以检测蛋白质表达。定量实时PCR进行检查以检查mRNA水平。进行了环己酰亚胺追逐测定法,以评估USP1的半衰期。共沉淀用于分析USP1的多泛素化。结果:CRC细胞中蛋白酶体抑制剂MG132增强了USP1蛋白稳定性。野生型USP1被MG132上调,但没有其催化突变体。此外,MG132也增强了USP1的多泛素化,这表明USP1通过泛素蛋白蛋白酶体途径降解。同时,我们证实ML323在CRC细胞中下调了USP1的表达,并且环己酰亚胺追逐测定也显示ML323降低了USP1蛋白质的稳定性。进一步的结果表明,MG132消除了ML323诱导的USP1下调和不稳定。此外,caspase抑制剂Z-VAD并未逆转USP1蛋白的不稳定,这进一步表明ML323诱导的USP1下调并不取决于CRC细胞中细胞死亡的影响。结论:我们的结果表明USP1是自动泛素化的,ML323通过CRC细胞中的泛素蛋白酶体途径不稳定USP1,为抗CRC药物的开发提供了针对USP1的理论基础。关键字:大肠癌,USP1,ML323

KSQ-4279:一种用于治疗具有同源重组缺陷癌症的第一类USP1抑制剂
•KSQ-4279是一种可逆的,具有ki = 1.2nm的USP1的变构抑制剂•ksq-4279绑定和未结合的USP1结构均已解决,揭示了诱导的拟合机构

关于临床USP1抑制剂作用机理的结构和生化见解,KSQ-4279
DNA损伤会触发介导修复的细胞信号级联。此信号在癌症中经常失调。介导该信号传导的蛋白质是治疗干预的潜在靶标。泛素特异性蛋白酶1(USP1)就是一个靶标,在临床试验中已经有小分子抑制剂。在这里,我们使用生化测定和冷冻电子显微镜(Cryo-EM)来研究临床USP1抑制剂KSQ-4279(RO7623066),并将其与已建立良好的工具化合物ML323进行比较。我们发现KSQ-4279与ML323的USP1同一隐性位点结合,但以微妙的方式破坏蛋白质结构。抑制剂结合使USP1的热稳定性大大提高,该抑制剂可以通过填充USP1中疏水隧道的抑制剂介导。我们的结果有助于理解分子水平USP1抑制剂的作用机理。
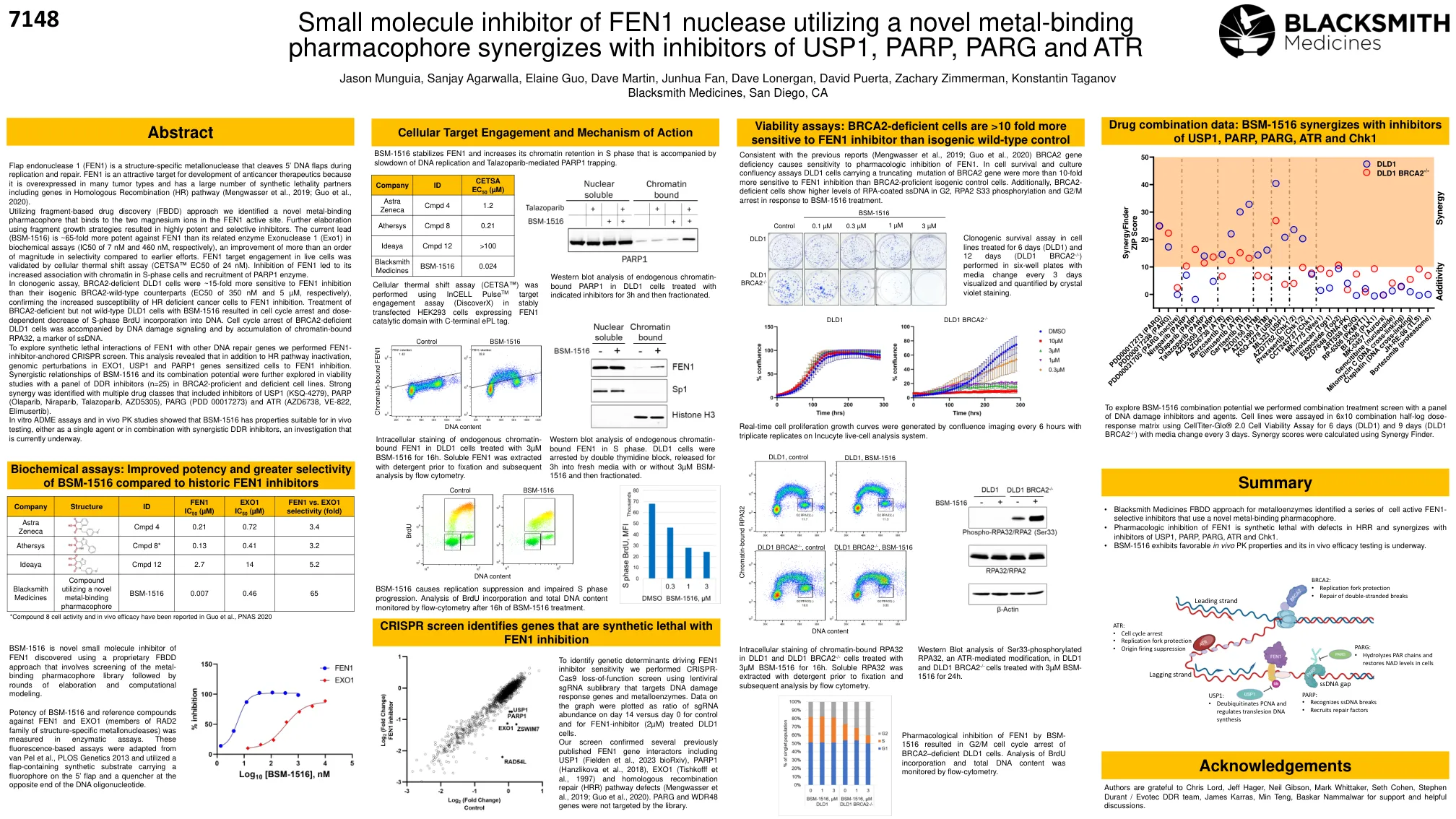
使用新型金属结合药效团与USP1,PARP,PARG和ATR抑制剂协同的Fen1核酸酶的小分子抑制剂
皮瓣核酸内切酶1(Fen1)是一种结构特异性的金属核酸酶,在复制和修复过程中切割5'DNA瓣。fen1是开发抗癌疗法的有吸引力的靶标,因为它在许多肿瘤类型中过表达,并且具有大量的合成致死性伴侣,包括同源重组基因(HR)途径(Mengwasser等,2019; Guo等,2020)。利用基于碎片的药物发现(FBDD)方法,我们确定了一种新型的金属结合药效团,该药效团与Fen1活性位点中的两个镁离子结合。使用碎片增长策略进一步阐述导致高度有效和选择性抑制剂。在生物化学测定中(分别为7 nm和460 nm的IC50),对FEN1的当前铅(BSM-1516)对FEN1的有效性比其相关酶外核酸酶1(EXO1)高65倍,与早期努力相比,改善了量的更大范围。fen1靶标在活细胞中的靶标参与通过细胞热偏移分析验证(CETSA
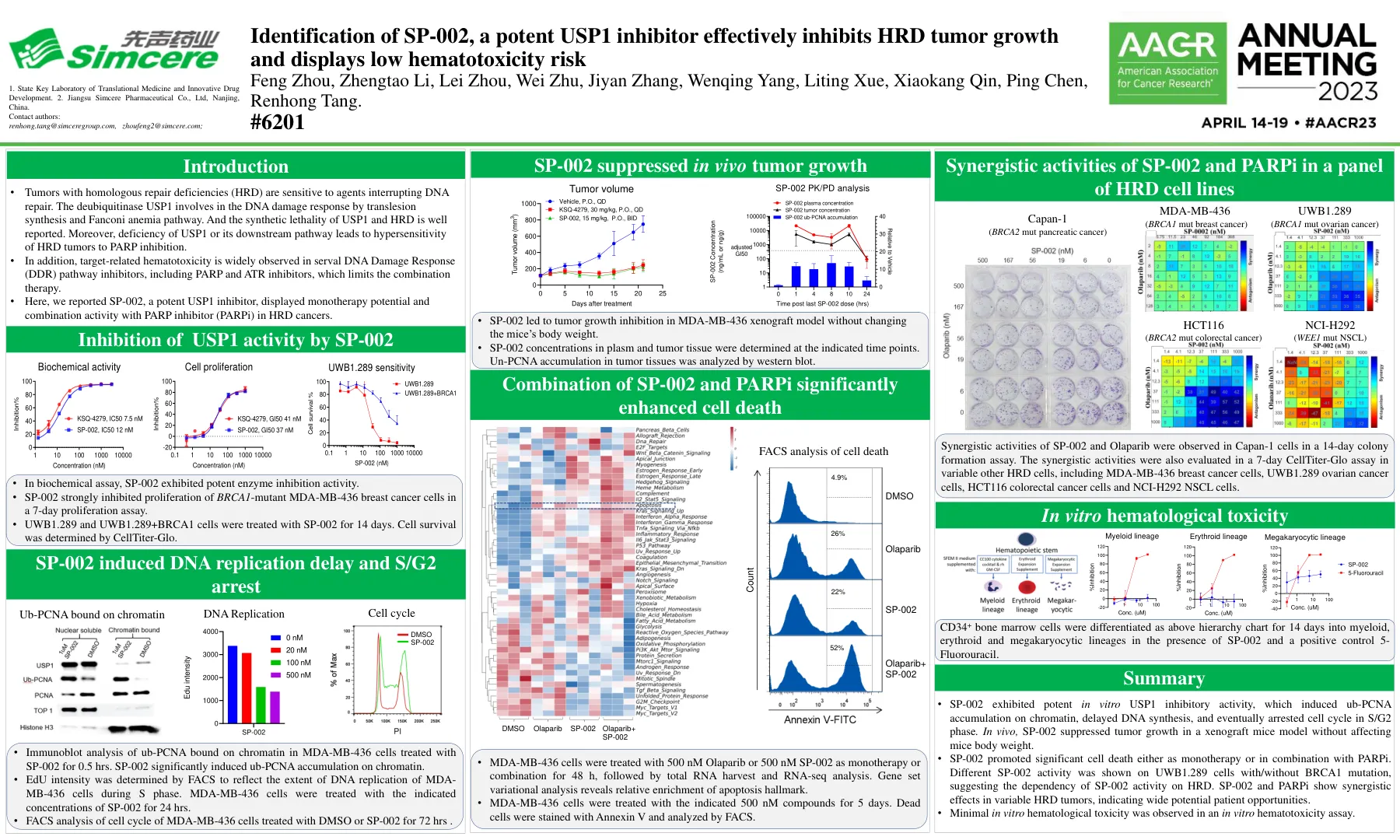
鉴定SP-002,有效的USP1抑制剂有效抑制HRD肿瘤的生长,并显示低血毒性风险风险王后,Zhengtao li,l
•具有同源修复缺陷(HRD)的肿瘤对中断DNA修复的药物敏感。去泛素酶USP1涉及Translesion合成和Fanconi贫血途径的DNA损伤反应。和USP1和HRD的合成致死性得到很好的报道。此外,USP1或其下游途径的缺乏会导致HRD肿瘤对PARP抑制过敏。•此外,与靶标相关的造血毒性被广泛观察到在包括PARP和ATR抑制剂在内的serval DNA损伤反应(DDR)途径抑制剂,这限制了联合疗法。•在这里,我们报道了SP-002是一种有效的USP1抑制剂,在HRD癌症中显示了与PARP抑制剂(PARPI)的单一疗法潜力和组合活性。

泛素化 PCNA 驱动 USP1 在癌症中的合成致死性 Antoine Simoneau、Justin L. Engel、Madhavi Bandi、Katherine Lazarides、Shangtao Liu、Samu
合成致死是一种遗传相互作用,指两个基因(但不是单独一个基因)丢失,会导致细胞死亡,并允许靶向疗法选择性地杀死肿瘤细胞,同时在很大程度上保护正常细胞。PARP 抑制剂获批用于治疗 BRCA1/2 突变癌症,这是合成致死概念的首个临床验证 (1)。鉴于 PARP 抑制剂的成功,人们对开发下一代合成致死癌症疗法产生了浓厚的兴趣。基于 CRISPR-Cas9 的功能基因组学的最新进展,加上对癌症遗传学知识的不断加深,正在加速针对癌症中新的遗传依赖性的靶向治疗。USP1 编码一种 785 个氨基酸的半胱氨酸蛋白酶,属于 USP 去泛素化酶家族 (2)。为了优化催化活性,USP1 与 UAF1 (2) 形成异二聚体复合物,UAF1 是一种含有 WD40 重复序列的蛋白质,也能刺激 USP46 和 USP12 (3)。 USP1 – UAF1 复合物使参与 DNA 损伤反应的几种底物去泛素化,包括单泛素化的 PCNA 和 FANCD2 (2, 4 – 6)。USP1 在跨损伤合成 (TLS) 和模板转换 (TS) DNA 损伤耐受过程中起着关键作用