XiaoMi-AI文件搜索系统
World File Search SystemZEB1

Zeb1和TMEJ在调节乳腺癌基因组稳定性
乳腺癌细胞经常在忠实的DNA修复基因中获取突变,例如BRCA降低的效率。此外,不准确的DNA修复途径的过表达也可能是癌症进展过程中遗传不稳定的起源。POLQ表达中的特定增益,编码参与theta介导的末端连接(TMEJ)的易于的DNA聚合酶theta(polθ)与特征突变签名有关。为了深入了解POLQ表达的机械调节,这篇评论介绍了有关Claudin-Low乳腺肿瘤亚型POLQ的调节的最新发现,这些调节特定地表达了参与上皮到 - 质质转变(EMT)(例如Zeb1)和诸如Zeb1和Paimic Abn in paimic abn的上皮性转变(EMT)的转录因子。

Zeb1保持长期的成年造血干细胞功能和耗尽造血的
新兴证据暗示上皮 - 间质转变转录因子ZEB1是造血干细胞(HSC)分化的关键调节剂。ZEB1是否调节HSC功能的长期维护仍然是一个空旷的问题。Using an inducible Mx-1-Cre mouse model that deletes condi- tional Zeb1 alleles in the adult hematopoietic system, we found that mice engineered to be de fi cient in Zeb1 for 32 weeks displayed expanded immunophenotypically de fi ned adult HSCs and multipotent progenitors associated with increased abundance of lineage-biased/balanced HSC subsets and augmented cell生存特征。在造血分化期间,持续的Zeb1损失增加了骨髓和脾脏中的B细胞,并减少了外周血中的单核细胞产生。在竞争性转移实验中,我们发现来自长期ZEB1缺失的成年小鼠的HSC在多列元素分化能力中显示出细胞自主缺陷。长期的Zeb1损失受干扰的髓质外造血作用,其特征是脾脏重量增加和脾细胞的矛盾降低,伴有HSC疲惫,谱系特异性缺陷,特异性缺陷,以及异常的,prelect的累积,诸如C-Kkit + CD16/32 + CD16/32 + Quertors的累积。ZEB1损失长达42周可以导致脾肿大和GR-1 + MAC-1 +细胞的积累,进一步支持这样一个观念,即Zeb1的长期表达抑制了PRELEUKEATIC活性。©2024 ISEH - 血液学和干细胞协会。由Elsevier Inc.出版因此,持续的Zeb1 de te骨会破坏体内HSC功能,并损害对耗尽造血的调节,对髓样肿瘤中Zeb1的肿瘤抑制功能有潜在的影响。这是CC下的开放式访问文章(http://creativecommons.org/licenses/4.0/)

EMT 转录因子 ZEB1 抑制乳腺癌中的致突变 POL u 介导的末端连接途径
摘要 ◥ 癌症发展的一个特征是获得基因组不稳定性,这是由于 DNA 损伤修复不准确造成的。在致癌应激诱导的双链断裂修复机制中,高度诱变的 theta 介导末端连接 (TMEJ) 通路已被证明在多种人类癌症中过表达,该通路需要由 POLQ 基因编码的 DNA 聚合酶 theta (POL q )。然而,人们对 TMEJ 的调节机制及其失调的后果知之甚少。在本研究中,我们结合生物信息学方法,探索乳腺癌国际联盟分子分类学和 Cancer Genome Atlas 数据库,并使用 CRISPR/Cas9 介导的 claudin-low 肿瘤细胞中锌指 E-box 结合同源框 1 (ZEB1) 的消耗,或在基底样肿瘤细胞(两种三阴性乳腺癌 (TNBC) 亚型)中强制表达 ZEB1,以证明 ZEB1 抑制
靶向CDK4克服EMT介导的肿瘤异质性和KRAS突变肺癌的治疗性
引言激活KRAS突变是肺癌中最常见的致癌事件之一,发生在约30%的肺腺癌患者中(1-3)。尽管鉴定出20年前的癌基因,并且为治疗这一子群而进行了重大努力,但5年的存活率仍然令人沮丧(4)。与EGFR - 突变肺癌不同,KRAS癌蛋白在很大程度上不可能,而最近的KRAS G12C等位基因除外(5,6)。MAPK途径的药理学抑制剂(例如MEK),例如selumetinib和trametinib,但临床前试验和临床试验表明对MEK抑制剂的反应不佳(7)。MEK抑制剂与常规化学疗法的结合并未证明无进展生存的额外好处(8)。 对MEK抑制剂的耐药性可能是固有的(从头),由于肿瘤细胞异质性或由于肿瘤进化而获得的作为对药理剂的适应性反应。 在任何一种情况下,都具有重编程的细胞机械上表型不同的肿瘤细胞亚群的存在使得难以有效消除更广泛的肿瘤细胞群。 为了解决这个问题,我们需要了解异质肿瘤内肿瘤细胞亚群的差异。 遗传上相同的肿瘤细胞具有进行转录重编程以激活替代生存途径并逃避治疗靶向的能力。 我们以前的研究强调了的依赖结合并未证明无进展生存的额外好处(8)。对MEK抑制剂的耐药性可能是固有的(从头),由于肿瘤细胞异质性或由于肿瘤进化而获得的作为对药理剂的适应性反应。在任何一种情况下,都具有重编程的细胞机械上表型不同的肿瘤细胞亚群的存在使得难以有效消除更广泛的肿瘤细胞群。为了解决这个问题,我们需要了解异质肿瘤内肿瘤细胞亚群的差异。遗传上相同的肿瘤细胞具有进行转录重编程以激活替代生存途径并逃避治疗靶向的能力。我们以前的研究强调了我们小组和其他人的研究表明,上皮 - 间充质转变(EMT)是在KRAS突变肺癌中发生的一种核心现象,这有助于细胞内肿瘤异质性,转移性的潜在增加,对药理学患者和贫穷的患者(9-111)(9-至11)。由KRAS和p53突变驱动的鼠肺癌模型概括了EMT介导的肿瘤细胞异质性,具有锌指E-Box结合HONEOBOX 1/miRNA-200(ZEB1/MIRNA-200(ZEB1/MIR-200)(ZEB1/MIR-200)(ZEB1/MIR-200))双重反馈回路在动态改变细胞概念(10)方面起着核心作用(10)。

DNMT3a 促进 LUAD 细胞增殖和转移...
背景:DNA甲基化模式的变化与肿瘤的发生发展密切相关,其中DNA甲基转移酶3α(DNMT3a)起着重要作用。但DNMT3a在肺腺癌(LUAD)中的作用和机制尚不清楚。本研究旨在探讨DNMT3a对LUAD细胞增殖和转移的潜在影响并探索其潜在的分子机制。方法:采用免疫组化和Kaplan-Meier生存分析方法探讨DNMT3a和组蛋白去乙酰化酶7(HDAC7)的表达与患者生存、预后及临床病理特征的关系。在体内和体外研究DNMT3a对LUAD细胞增殖和转移的影响。本研究采用重组慢病毒介导的体外基因过表达或敲减、蛋白质印迹法、定量实时聚合酶链式反应(qRT-PCR)等方法,阐明DNMT3a促进LUAD细胞增殖和转移的潜在分子机制。结果:DNMT3a或HDAC7高表达与LUAD患者预后不良、AJCC第8版分期高、肿瘤分化差呈正相关,DNMT3a/HDAC7共同低表达的LUAD患者预后最差。DNMT3a上调可以通过上调HDAC7,进一步激活下游介质ZEB1和c-Myc的表达,促进LUAD细胞增殖和转移。相反,HDAC7 的过表达逆转了由 DNMT3a 下调介导的肿瘤生长和转移的减弱以及 c-Myc 和 ZEB1 表达的抑制,进一步表明 LUAD 中 DNMT3a 和 HDAC7 之间存在正反馈调节。结论:我们的研究结果首次证实,DNMT3a 通过上调 HDAC7 并进一步诱导 ZEB1 和 c-Myc 上调,充当诱导 LUAD 恶性进展的肿瘤启动子。靶向 DNMT3a 和 HDAC7 可能是 LUAD 的一种有前途的治疗策略。
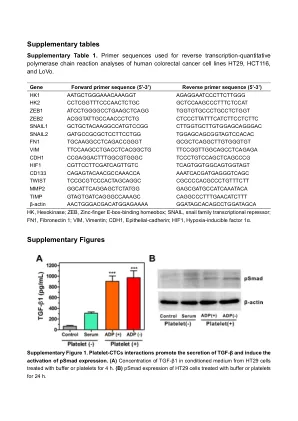
补充表
ctctttctctctc nail1 gctgctgtgtgtgtgtgtgtgtgtgtgtgtgtgtgtgtgtggtggggggghts snal2 TGCAAGCCTCGGGGTGTGTGGTGGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGCCCCCCCCCCCCCGCTGGTGGCTGGTGTGTGTGTGGTGH1 CDH1 CDGGTGGGGC TCCTGTCCAGCCCCGCCGCCCGCGCGCGCCGCGGS CGTTCTCTCTGTGTC TCAGTGGGGGGGGGTGGTGTGTGTGTs CD133 CAGTACCAACCAAACCIGITCIGITITIs GAGCIGITING TIMP GTAGGGGGGGGGGGGGGGGGGCAGC caggccttttgttttttttttttttttttttttttsβ-肌动蛋白actghtgacity gtatagcagcatgityctctttctctctc nail1 gctgctgtgtgtgtgtgtgtgtgtgtgtgtgtgtgtgtgtggtggggggghts snal2 TGCAAGCCTCGGGGTGTGTGGTGGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGCCCCCCCCCCCCCGCTGGTGGCTGGTGTGTGTGTGGTGH1 CDH1 CDGGTGGGGC TCCTGTCCAGCCCCGCCGCCCGCGCGCGCCGCGGS CGTTCTCTCTGTGTC TCAGTGGGGGGGGGTGGTGTGTGTGTs CD133 CAGTACCAACCAAACCIGITCIGITITIs GAGCIGITING TIMP GTAGGGGGGGGGGGGGGGGGGCAGC caggccttttgttttttttttttttttttttttttsβ-肌动蛋白actghtgacity gtatagcagcatgity

黑色素瘤中增殖性侵入性可塑性和IFNγ信号的动态建模揭示了PD-L1表达异质性的机制
黑色素瘤细胞的抽象背景表型异质性有助于耐药性,增加的转移和免疫逃避性疾病。各自的机制已被据报道,以塑造广泛的肿瘤内和肿瘤间表型异质性,例如IFNγ信号传导和对侵入性过渡的增殖,但是它们的串扰如何影响肿瘤的进展仍然很大程度上难以捉摸。在这里,我们将动态系统建模与散装和单细胞水平的转录组数据分析整合在一起,以研究黑色素瘤表型异质性背后的基本机制及其对适应靶向治疗和免疫检查点抑制剂的影响。我们构建了一个最小的核心监管网络,该网络涉及与此过程有关的转录因子,并确定该网络启用的表型景观中的多个“吸引子”。在三种黑色素瘤细胞系(Malme3,SK-MEL-5和A375)中,通过IFNγ信号传导和增生对浸润性转变对PD-L1的协同控制进行了模型预测。结果我们证明,包括MITF,SOX10,SOX9,JUN和ZEB1的调节网络的新兴动态可以概括有关多种表型共存的实验观察结果(增殖性,神经CREST,类似于神经crest,类似于Invasive),以及可转化的细胞检查和响应的响应,包括对响应的响应,并在响应中进行了响应,并在响应中置于某些响应中,并在构成方面构成了对响应的响应。这些表型具有不同水平的PD-L1,在免疫抑制中驱动异质性。PD-L1中的这种异质性可以通过这些调节剂与IFNγ信号的组合动力学加剧。我们关于黑色素瘤细胞逃避靶向治疗和免疫检查点抑制剂的侵入性转变和PD-L1水平的变化的模型预测在来自体外和体内实验的多个RNA-SEQ数据集中得到了验证。结论我们的校准动力学模型提供了一个测试组合疗法的平台,并为转移性黑色素瘤的治疗提供了理性的途径。可以利用对PD-L1表达,侵入性过渡和IFNγ信号传导增殖的串扰的改进理解,以改善对治疗耐药和转移性黑色素瘤的临床管理。