XiaoMi-AI文件搜索系统
World File Search Systemcber
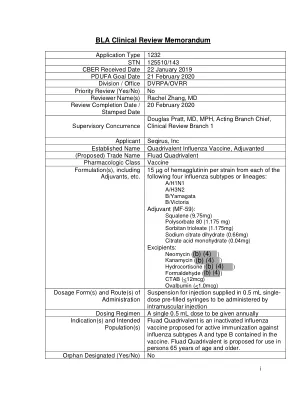
四价氟化物
AE 不良事件 AESI 特别关注的不良事件 aQIV 佐剂四价流感疫苗 AR 不良反应 aTIV 佐剂三价流感疫苗 BIMO CBER 生物研究监测 BLA 生物制品许可证申请 CBER 生物制品评价与研究中心 CFR 联邦法规 CI 置信区间 CMC 化学、制造与控制 CRF 病例报告表 CSR 临床研究报告 FAS 完整分析集 FDA 食品药品管理局 GMT 几何平均滴度 HA 血凝素 HI 血凝抑制 ICH 国际协调会议 ILI 流感样疾病 LL 下限 MedDRA 监管活动医学词典 NOCD 新发慢性病 OBE 生物统计学和流行病学办公室 OVRR 疫苗研究与审查办公室 PeRC 儿科审查委员会 PI 说明书 PMC 上市后承诺 PMR 上市后要求 PPS 按照方案集PREA 儿科研究公平法案 PT 首选术语 QIV 四价流感疫苗 RT-PCR 逆转录聚合酶链反应 SAE 严重不良事件 sBLA 补充生物制品许可申请 SCR 血清转化率 SOC 系统器官分类 STN 提交追踪编号 US 美国 WHO 世界卫生组织

疫苗试验的数据标准一致性检查
• 一致性检查旨在支持对研究 SDTM 数据集中的反应原性数据的审查。 • SAS 检查套件可以在试验期间的不同时间点执行,这对研究非常有益。 • 在研究设置时已经可以检测到 SDTM 映射中的不一致,并且可以在数据库锁定之前执行最终运行,以确保研究 SDTM 数据足以提交给 FDA CBER OVRR。 • 目前,一致性检查侧重于反应原性数据。未来可能会包括对 FDA 指南中描述的其他类型数据的检查。

1 CNMC/FDA-CBER儿科传染病...
CNMC/FDA-CBER儿科传染病研究生课程背景:儿童国家医疗中心(CNMC)的小儿传染病和生物学评估与研究中心(FDA-CBER)的小儿药物管理局(FDA-CBER)分子培训计划。所有临床活动和培训都发生在CNMC,而FDA-CBER为美国儿科学委员会(ABP)培训要求的资助,指导和物理环境,以进行学术活动,以进行专业认证。fda-cber研究员在CBER疫苗,疫苗和相关产品应用程序(DVRPA)的CBER办公室内担任医务人员。本文档描述了FDA-CBER奖学金计划部分的正式课程。成功完成FDA-CBER奖学金课程的目标和目标(由ACGME核心能力分类):患者护理/监管实践•受训者必须在制定疫苗和相关产品的风险效率分析时练习合理的医疗决策。•学员必须表现出精通清晰,简洁且经过良好经验的书面临床评论。•受训者必须证明熟练的监管计算机系统。医学/监管知识•受训者必须证明对FDA调节过程的开发和许可程序和相关产品的许可,包括:1。临床前和临床开发的阶段2。许可过程3.销售后的问题。销售后问题•受训者必须熟悉FDA在中心,办公室,办公室和部门的功能组织。•学员必须表现出对适用于审查程序的法律和法规的工作理解。•学员必须表现出对监管审查团队组成和职责的理解。•学员必须对良好的科学临床试验设计和道德临床试验的必要组成部分表示赞赏。•学员必须证明对人类对疫苗抗原和辅助因素的免疫反应的一般理解,以及对当前实验室工具和用于测量免疫反应的技术的一般理解。学员还必须能够定义和区分保护和保护的替代物,并描述如何将这些概念应用于疫苗和相关产品的调节。•学员必须对安全性,纯度,效能和功效的概念进行一般的了解,因为他们适用于疫苗和相关产品。

Quantitative Approaches to Select Dosages for Clinical Trials
• OCE: Rick Pazdur, Marc Theoret • Leads: Atik Rahman, Mirat Shah • RPM: Pam Balcazar • Pharmacology/Toxicology: Haleh Saber, Matthew Thompson • Clinical Pharmacology: Brian Booth, Lanre Okusanya, Stacy Shord • Pharmacometrics: Jiang Liu, Hao Zhu • OCP Policy: Raj Madabushi • Clinical: Brian Heiss, Jennifer Gao, Gwynn Ison, Elizabeth Duke, Shruti Gandhy, Cara Rabik, Pam Seam • Safety: Abhi Nair • Biostatistics: Joyce Cheng, Jonathon Vallejo, Gary Rosner • CBER: Lianne Wu, Xiaofei Wang • Analysts: Alex Akalu, Susan Jenney

行业,调查人员和其他利益相关者的指南
1,该指南是由医疗政策办公室在药物评估与研究中心(CDER)与生物学评估与研究中心合作(CBER),设备和放射学健康中心(CDRH)和卓越肿瘤学中心(OCE)(OCE)(FDA)(FDA)的肿瘤学中心(OCE)。2个单词和短语。3虽然本指南的重点是用于临床研究中用于远程数据获取的DHT,但当试验参与者在试验地点使用DHT时,本指南中的建议可能适用(例如,在临床中审查了连续的葡萄糖监测器)。4有关FDA的临床研究或研究的监管定义,请参见21 CFR 50.3(c),56.102(c),312.3(b)和812.3(h)。为了本指南,临床试验和临床研究术语可互换使用。5就本指南而言,所有提及医学产品的意思是人类药物和生物产品,医疗设备和组合产品(请参见21 CFR 3.2(e)),该产品受CDER,CBER或CDRH的调节。6这可能包括人工智能(AI)启用的软件。7就本指南而言,对于任何给定的DHT,术语函数是DHT在临床研究中的独特目的,这可能是预期使用或预期使用DHT的子集。DHT可以具有多个功能,可能是设备功能也可能不是设备功能。在考虑FDA的多功能设备产品的监管方法和政策时,请参阅行业和FDA员工多功能设备的指南

审查备忘录 - Moderna COVID-19 疫苗
审查备忘录 日期:2021 年 11 月 19 日 收件人:文件 来自:David Cho 博士(CBER/OD) 发件人:Peter Marks 医学博士、博士(CBER/OD) 申请人名称:ModernaTX, Inc. 申请编号:EUA 27073 产品:Moderna COVID-19 疫苗 主题:CBER 对 18 岁及以上个人在进行 COVID-19 初级免疫系列后接种的 Moderna COVID-19 疫苗(0.25 毫升)加强剂量的评估 本备忘录总结、审查和建议 Moderna 于 2021 年 11 月 11 日提交的修改其 COVID-19 疫苗紧急使用授权 (EUA) 的提案,以授权在对 18 岁及以上的个人进行 COVID-19 初级免疫系列后接种加强剂量。执行摘要 Moderna 已对 EUA 27073 提出修订建议,以包括在对至少 18 岁的个人进行 COVID-19 基本免疫系列后注射加强剂。参考 2020 年 12 月 18 日发布的 Moderna COVID-19 疫苗 EUA,该 EUA 基于约 30,000 名参与者的安慰剂对照随机试验描述了该疫苗的安全性和有效性。Moderna 目前的授权适应症是用于主动免疫,以预防 18 岁及以上人群中由严重急性呼吸综合征冠状病毒 2 (SARS-CoV-2) 引起的 2019 冠状病毒病 (COVID-19)。2021 年 8 月 12 日,FDA 修订了 Moderna COVID-19 疫苗 EUA,授权向某些免疫功能低下的个人给予额外剂量。 2021 年 10 月 20 日,FDA 修订了 Moderna COVID-19 疫苗 EUA,授权在完成主要系列接种后至少 6 个月为 65 岁及以上的个人、18 至 64 岁患严重 COVID-19 风险较高的个人以及 18 至 64 岁经常在机构或职业中接触 SARS-CoV-2 的个人接种一剂 Moderna COVID-19 疫苗加强剂。同样在 2021 年 10 月 20 日,FDA 授权使用目前可用的(即 FDA 授权或批准的)COVID-19 疫苗的异源加强剂。Moderna COVID-19 加强剂是主要系列接种剂量的一半。根据修订后的效益风险评估,Moderna 请求修改其 COVID-19 疫苗的 EUA,以授权在主要 COVID-19 接种后至少 6 个月接种加强剂
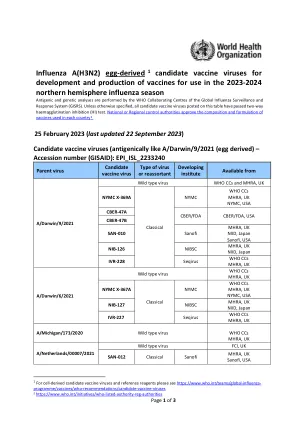
甲型流感(H3N2)1 鸡蛋衍生候选疫苗...
AS445 TGA,澳大利亚 2023AH3-1* NIID,日本 *新试剂以蓝色显示 上述效力测试试剂是在制造商支持下开发的,并由全球流感监测和应对系统 (GISRS) 的世卫组织基本监管实验室 (ERL) 校准。有关试剂订单/信息的 ERL 联系方式: CBER:CBERshippingrequests@fda.hhs.gov MHRA:standards@nibsc.org 或 enquiries@nibsc.org NIID:flu-vaccine@nih.go.jp TGA:influenza.reagents@health.gov.au 有关其他候选疫苗病毒和效力测试试剂,请访问 https://www.who.int/teams/global-influenza-programme/vaccines/who-recommendations/candidate-vaccine-viruses 如有一般咨询,请联系 gisrs-whohq@who.int

药品和生物制品外部对照试验的设计和实施注意事项
指导草案 本指导文件仅供评论之用。有关本草案的评论和建议应在《联邦公报》上公布指导草案发布通知后 90 天内提交。请将电子评论提交至 https://www.regulations.gov。请将书面评论提交至食品药品管理局卷宗管理人员(HFA-305),地址:5630 Fishers Lane, Rm. 1061, Rockville, MD 20852。所有评论均应注明《联邦公报》上公布的发布通知中所列的卷宗编号。如对本草案有任何疑问,请联系 (CDER) Dianne Paraoan,电话:301-796-2500,或 (CBER) 沟通、宣传和发展办公室,电话:800-835-4709 或 240-402-8010。

注射产品中可见颗粒的检测
指导草案 本指导文件仅供评论之用。有关本草案的评论和建议应在《联邦公报》上公布指导草案发布通知后 60 天内提交。请将电子评论发送至 https://www.regulations.gov。请将书面评论发送至食品药品管理局卷宗管理人员(HFA-305),地址:5630 Fishers Lane, Rm. 1061, Rockville, MD 20852。所有评论均应注明在《联邦公报》上公布的发布通知中所列的卷宗编号。 如对本草案有任何疑问,请联系 (CDER) Eric Dong 240-402-4172;(CBER) 沟通、宣传和发展办公室,800-835-4709 或 240-402-8010;或 (CVM) AskCVM@fda.hhs.gov。

药物和生物产品开发的主协议
指南草案此指南文件仅用于评论目的。有关此文件草案的评论和建议应在联邦公报发表后的60天内提交通知,宣布指导草案的可用性。将电子评论提交https://www.regulations.gov。向码头管理人员(HFA-305)提交书面评论,食品和药物管理局,5630 Fishers Lane,RM。1061,Rockville,MD 20852。应将所有评论与在联邦登记册上发布的可用性通知书中列出的案卷号一起识别。有关此文件草案的疑问,请致电301-796-2055与Scott N. Goldie联系,或(CBER)通讯,外展与发展办公室,800-835-4709或240-402-8010.