XiaoMi-AI文件搜索系统
World File Search Systemdystrophy

Fuchs营养不良
在大量的眼睛和耳朵上,我们是美国Fuchs营养不良症的最高量治疗计划之一。在过去的十年中,我们的研究人员开发了研究计划来了解疾病机制并开发了新颖的手术技术,从而减少了角膜移植后的恢复时间。虽然对Fuchs营养不良的治疗曾经需要进行全厚的角膜移植,但大量的眼睛和耳外科医生采用了内皮性角膜成形术,First DSAEK,然后是DMEK,以治疗紫红色的营养不良,并具有微不足道的手术性手术干预措施。dmek是一种最先进的技术,具有减少的移植排斥,更快的恢复时间和更好的视力结果。另一种创新的外科手术方法,只有descemet的剥离(DSO),该方法可以消除患病的内皮细胞,而无需供体组织植入,以大量眼睛和耳朵的培养基治疗而无需移植的Fuchs营养不良而无需移植。

facioscapulohumeral肌肉营养不良
当脊柱周围的肌肉削弱时,将色谱柱退出对准。未对准会导致脊柱侧弯,脊柱弯曲到侧面。当背部的肌肉较弱时,个体难以直立站立,脊柱在下背部向内弯曲,被称为Lordosis。脊柱侧弯和脊柱障碍可能在FSHD中是温和的。

婴儿神经轴索营养不良症
疾病名称:婴儿神经轴索营养不良 ICD 10:G23.0 同义词:INAD、NBIA2、磷脂酶 A2 相关神经变性 (PLAN)、Seitelberger 病、伴有脑铁沉积的神经变性 A 疾病摘要:婴儿神经轴索营养不良 (INAD) 是一种与 PLA2G6 基因突变相关的神经变性疾病。它是继泛酸激酶相关神经变性 (PKAN,以前称为 Hallervorden-Spatz 病) 之后第二常见的伴有脑铁沉积 (NBIA) 的神经变性类型。INAD 以常染色体隐性方式遗传。 PLA2G6 编码钙非依赖性磷脂酶,与婴儿神经轴突营养不良 (INAD)、非典型神经轴突营养不良 (NAD) 和肌张力障碍-帕金森病有关。PLA2G6 表达于线粒体健康,并保护线粒体健康。它对膜稳态和钙信号传导也很重要。INAD 的组织学特征是轴突球体。表型上,INAD 的特征是心理运动退化,发病早于 6 个月至 3 岁之间。肌张力低下发生早,伴有严重虚弱,可能被痉挛取代。许多 INAD 患者还会出现进行性痴呆。患者通常在 10 岁之前因呼吸系统并发症死亡。患者可能因延髓功能障碍而接受胃造口管和气管切开术,有些患者可能需要手术矫正脊柱侧弯以改善呼吸状况。 INAD 患者的主要麻醉问题是他们术前呼吸状况不佳,这是由于气道清除和呼吸力学较差导致的,因此通常需要术后插管。

关于Duchenne肌肉营养不良症的评论
- 仪性患者在NSAA的6MWT中表现出改善。- 上肢测试,强度和fa tigue耐药性和氨基胶质(EMG)参数的改良性能。- 不需要免疫抑制。- 索引在目标器官中的dys-trophin。

肌发育症的基因检测
描述/背景肌肉营养不良是指30多种遗传性疾病,这些疾病会导致渐进的肌肉无力和肌肉丧失。肌发育症(DM)是一种肌肉营养不良的一种,具有2种形式,1型(DM1)和2型(DM2)。这是成人发作的肌肉营养不良的最常见形式。1 DM1 DM1,也称为Steinert病,估计会影响全球20,000名个人中的1个。流行率似乎是区域性的,可能高达10,000(冰岛)中的1个,达到100,000分之一(在日本的某些地区)。它是以常染色体主导方式继承的。dm1是由位于染色体19。ctg重复长度最多34被认为是正常的。重复长度在35至49之间,尽管异常,但不会导致症状表达;但是,这些人的孩子有更大重复的风险增加(预期)。使用DM1,重复可能超过5,000。2,3 CTG重复扩张的长度与疾病的严重程度中等相关。DM1是一种多系统疾病,可能会影响大脑,骨骼和光滑的肌肉,眼睛,心脏,胃肠道,肺部和内分泌系统。它是最可变的遗传性人类疾病之一,因为该表达范围从无症状成年人到严重影响的新生儿。临床发现已分为3种表型;但是,它们之间没有绝对的区别,因为它们更像是连续体。- 轻度DM1(成人发言)是最普遍的形式,可能包括过早的白内障,秃发和轻度肌瘤;寿命是正常的。- 经典DM1(成人发言)可能包括白内障;远端弱点涉及腿的背屈者和手臂的长手指屈肌;手,脖子和脸部的肌肉动物;

福山先天性肌肉营养不良
•saito K.福山先天性肌肉营养不良。2006年1月26日[更新于2019年7月3日]。in:Adam MP,Feldman J,Mirzaa GM,Pagon RA,Wallace SE,Amemiya A,Editors.genereviews(R)[Internet]。西雅图(WA):西雅图华盛顿大学; 1993-2025。 可从http://www.ncbi.nlm.nih.gov/books/nbk1206/引用(https://pubmed.ncbi.nlm.nih.gov/20301385) Tachikawa M,Wang F,Nagai Y,Taniguchi K,Taniguchi M,Sunada Y,Terashima T,Endo T,Matsumura K.Fukuyama-type先天性肌营养不良症(FCMD)Andalpha-delpha-dyalpha- dystroglycanopathy。 Anmit Anom(Kyoto)。 2003 Jun; 43(2):97-104。 doi:10.1111/j。 1741-4520.2003.tb01033.x。 Citation on PubMed (https://pubmed.ncbi.nlm.nih.gov/12 893968) • Yoshioka M, Higuchi Y, Fujii T, Aiba H, Toda T. Seizure-genotype relationshipin Fukuyama-type congenital muscular dystrophy. 大脑开发。 2008 JAN; 30(1):59-67.DOI:10.1016/j.braindev.2007.05.012。 Epub 2007年6月26日。 引用PubMed(https://pu bmed.ncbi.nlm.nih.gov/17597323)•Yoshioka M,BurokiS。 Am J Med Genet。 1994年11月15日; 53(3):245-50。doi:10.1002/ajmg.1320530309。 PubMed的引用(https://pubmed.ncbi.nlm.nih .gov/7856660)西雅图(WA):西雅图华盛顿大学; 1993-2025。可从http://www.ncbi.nlm.nih.gov/books/nbk1206/引用(https://pubmed.ncbi.nlm.nih.gov/20301385) Tachikawa M,Wang F,Nagai Y,Taniguchi K,Taniguchi M,Sunada Y,Terashima T,Endo T,Matsumura K.Fukuyama-type先天性肌营养不良症(FCMD)Andalpha-delpha-dyalpha- dystroglycanopathy。Anmit Anom(Kyoto)。2003 Jun; 43(2):97-104。 doi:10.1111/j。 1741-4520.2003.tb01033.x。 Citation on PubMed (https://pubmed.ncbi.nlm.nih.gov/12 893968) • Yoshioka M, Higuchi Y, Fujii T, Aiba H, Toda T. Seizure-genotype relationshipin Fukuyama-type congenital muscular dystrophy. 大脑开发。 2008 JAN; 30(1):59-67.DOI:10.1016/j.braindev.2007.05.012。 Epub 2007年6月26日。 引用PubMed(https://pu bmed.ncbi.nlm.nih.gov/17597323)•Yoshioka M,BurokiS。 Am J Med Genet。 1994年11月15日; 53(3):245-50。doi:10.1002/ajmg.1320530309。 PubMed的引用(https://pubmed.ncbi.nlm.nih .gov/7856660)2003 Jun; 43(2):97-104。 doi:10.1111/j。1741-4520.2003.tb01033.x。Citation on PubMed (https://pubmed.ncbi.nlm.nih.gov/12 893968) • Yoshioka M, Higuchi Y, Fujii T, Aiba H, Toda T. Seizure-genotype relationshipin Fukuyama-type congenital muscular dystrophy.大脑开发。2008 JAN; 30(1):59-67.DOI:10.1016/j.braindev.2007.05.012。 Epub 2007年6月26日。 引用PubMed(https://pu bmed.ncbi.nlm.nih.gov/17597323)•Yoshioka M,BurokiS。 Am J Med Genet。 1994年11月15日; 53(3):245-50。doi:10.1002/ajmg.1320530309。 PubMed的引用(https://pubmed.ncbi.nlm.nih .gov/7856660)2008 JAN; 30(1):59-67.DOI:10.1016/j.braindev.2007.05.012。Epub 2007年6月26日。引用PubMed(https://pu bmed.ncbi.nlm.nih.gov/17597323)•Yoshioka M,BurokiS。Am J Med Genet。1994年11月15日; 53(3):245-50。doi:10.1002/ajmg.1320530309。PubMed的引用(https://pubmed.ncbi.nlm.nih .gov/7856660)

针对杜氏肌营养不良症
约翰·基里安:如果说得通的话,感觉就像昨天和很久以前一样。约翰·基里安这样描述他儿子得到改变人生的诊断结果的那一天。约翰·基里安:我还记得当时的具体情况,听到的那些话,这是你一生中永远不会忘记的事情之一。那是 17 年前的事了。约翰·基里安:萨姆在三岁时被诊断出患有癌症。对于约翰和他的妻子斯蒂芬妮来说,17 年来他们不知道儿子的未来会怎样。十七个生日……他们每个人都希望萨姆能活到下一个生日。迹象是存在的。但它们很微妙。萨姆在蹒跚学步时,发育情况与其他孩子不同:约翰·基里安:他只会坐,不会在正确的时间站起来。他是我们四个孩子中的第四个。所以我们非常了解孩子在什么年龄的表现。而且,我们也知道。嗯,孩子们发育的速度不同。所以我们并不太担心。甚至连萨姆的儿科医生一开始都没有发现任何问题。后来有一天,他们一家在家附近的公园玩耍时,萨姆摔断了腿。约翰·基利安:这就是我们最终得到的诊断结果。他的腿上打了六个星期的石膏,恢复起来很困难。我们去找了一位理疗师,试图为他寻求帮助。最后,她告诉我的妻子,“嘿,我觉得萨姆可能患有肌肉萎缩症。”大家好,我是乔丹·加斯-普雷,南加州大学健康新闻中心的成员。这是彭博媒体工作室和 Vertex Pharmaceuticals 的播客《针对最棘手的疾病》。
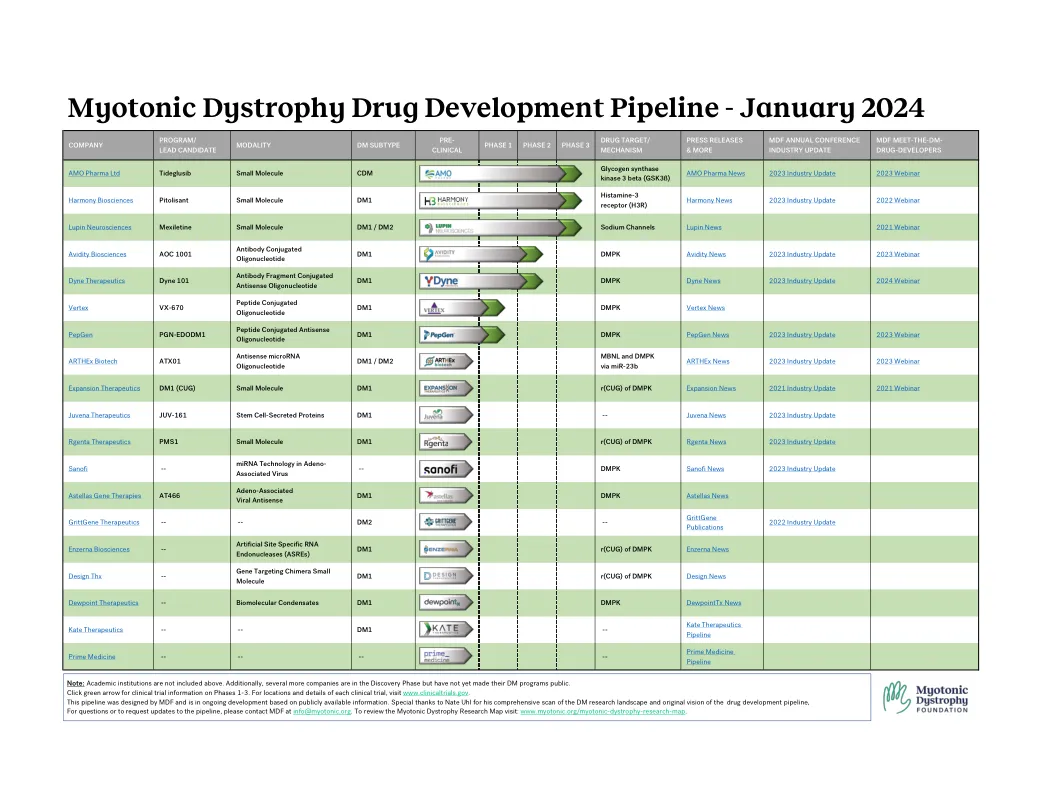
肌发育症药物开发管道
注意:上面不包括学术机构。此外,还有更多公司处于发现阶段,但尚未公开其DM计划。单击绿色箭头以获取有关阶段1-3的临床试验信息。有关每个临床试验的位置和细节,请访问www.clinicaltrials.gov。该管道是由MDF设计的,并且正在基于公开信息的持续开发。特别感谢Nate UHL对DM研究景观的全面扫描和药物开发管道的原始愿景,有关问题或要求对管道的更新,请通过info@myotonic.org与MDF联系。要查看肌发育症研究地图访问:www.myotonic.org/myotonic-dystrophy-research-map。

Duchenne肌肉营养不良中的惊吓反应
。cc-by-nc-nd 4.0国际许可证可永久提供。是作者/资助者,他已授予Medrxiv的许可证,以显示预印本(未通过同行评审证明)预印版本的版权所有者此版本发布于2021年9月22日。 https://doi.org/10.1101/2021.09.16.21263132 doi:medrxiv preprint

Stargardt 黄斑营养不良症及其治疗方法
摘要 斯塔加特黄斑营养不良症(Stargardt 病;STGD1;OMIM 248200)是最常见的遗传性黄斑营养不良症。STGD1 是一种常染色体隐性遗传病,由大 ABCA4 基因(OMIM 601691)中的多个致病序列变异引起。在理解临床和分子特征以及潜在病理生理学方面取得了重大进展,导致许多已完成、正在进行和计划中的新疗法人体临床试验。本简明综述的目的是描述(1)该疾病的详细表型和基因型特征、多模态成像发现、疾病的自然史和发病机制,(2)多种研究途径和治疗干预,包括药理学、细胞疗法和多种类型的基因疗法,这些疗法已经或正在研究中,以及(3)旨在通过替换整个 6.8 kb ABCA4 开放阅读框来治疗 STGD1 的激动人心的新型治疗方法。