XiaoMi-AI文件搜索系统
World File Search Systemdystrophy

咽喉肌营养不良研究和治疗更新
研究人员正在研究OPMD的疾病改良治疗方法。BB-301的1B/2A期临床试验是一种与腺相关的病毒载体分割的基因治疗,最近对其第一位患者施加了。研究性药物旨在沉默和替代突变的多A结合蛋白核-1(PABPN1)基因。
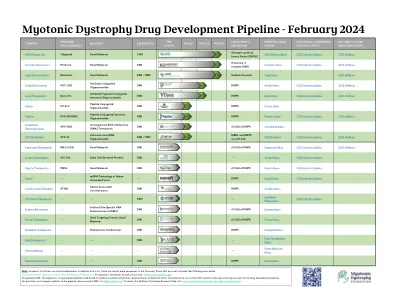
MDF 强直性肌营养不良症药物研发管线
注:以上不包括学术机构。除此列表外,还有几家处于发现阶段但尚未公开其 DM 计划的公司。单击上方的绿色箭头可查看第 1-3 阶段的临床试验信息。有关每个临床试验的地点和详细信息,请访问 www.clinicaltrials.gov 。此管道由 MDF 设计,正在持续开发中,并基于公开信息。特别感谢 Nate Uhl 对 DM 研究前景的全面审视以及对药物开发管道的原创愿景。如有疑问或需要管道更新,请联系 MDF,邮箱地址为 info@myotonic.org 。要查看强直性肌营养不良症研究地图,请访问:www.myotonic.org/myotonic-dystrophy-research-map 。

适合Duchenne肌肉营养不良症的合适动物模型
引言Duchenne肌营养不良症(DMD)是由编码细胞内蛋白质肌营养不良蛋白的基因突变引起的,是一种严重的X染色体染色体连接疾病,其特征是渐进的肌肉无力和变性。除了特征良好的骨骼肌病理学外,DMD还与相关的心脏并发症有关(Shirokova和Niggli,2013; Spurney,2011)。在其中,心律不齐和扩张的心肌病的发展极大地有助于与该疾病伴随的发病率和死亡率。在DMD背景下,导致心脏并发症的机制在很大程度上未知,这增加了对DMD动物模型的基础研究工作的需求。在使用的DMD动物模型中(McGreevy等,2015; Wells,2018),MDX小鼠是最著名的,最广泛使用的。它在鼠DMD基因的外显子23中具有过早的停止突变,因此未能翻译功能性全长肌营养不良蛋白(Sicinski等,1989)。尽管MDX小鼠是DMD的有用的遗传和生化模型,但仅部分模仿了人类疾病。因此,与DMD患者相比,MDX小鼠的寿命略有缩短,并且没有显示出明显的肌肉营养不良症状(Grady等,1997; Gutpell等,2015)。此外,MDX小鼠的心脏异常仅出现晚期(Quinlan等,2004),与DMD患者发生的心肌病相比是温和的(Grady等,1997; Janssen等,2005)。这质疑该动物模型研究心脏病表型的适用性。2014年,Larcher及其同事使用转录激活剂样效应子核酸酶靶向DMD基因的外显子23的发展肌营养不良蛋白缺陷型大鼠的发展(Larcher等,2014)。在这些DMD MDX大鼠中,心肌受坏死和纤维化的影响,并显示出进行性扩张性心肌病的迹象。超声心动图显示出明显的同心重塑和舒张功能的改变。基于这些发现,作者认为,DMD MDX大鼠中心脏病表型在DMD患者中观察到的,并且该动物模型可能适用于临床前DMD研究(Larcher等,2014)。该研究的弱点 - 实际关注骨骼肌肉 - 是DMD MDX大鼠的心脏病表型没有详细表征。例如,超声心动图仅对3个月大但不老的大鼠进行。此外,作者(Larcher等人,2014年)没有研究可能发生的血管并发症,例如增强的动脉僵硬度(Ryan等,2017)和内皮细胞(EC)功能障碍(Miike等,1987),这也可能有助于DMD患者的心脏病概念型的发展。最后,在细胞水平上的功能研究(即dmd MDX心肌细胞)尚未进行。考虑到缺乏证据,本研究的目的是提供处理编辑器的详细表征:Monica J.正义获得了2020年10月8日; 2020年12月23日接受

先天性强直性肌营养不良症的超声新发现
摘要 目的 通过超声检查,可以有限地诊断先天性强直性肌营养不良 (CDM),特别是在没有强直性肌营养不良 (DM) 家族史的情况下。我们回顾了 CDM 病例以寻找独特的产前发现。研究设计 一系列单中心病例,其中胎儿患有 CMD,具有特征性的产前发现,并在出生后得到证实。结果 四例产前或产后诊断为 CDM 的胎儿在宫内出现大头畸形。虽然头部测量结果在妊娠中期之前与胎龄相符,但妊娠晚期头围和双顶径均比平均值高出 >2 个标准差 (SD)。腹部和股骨测量结果与妊娠期相符。出生后,所有胎儿的枕额周长均比平均值高出 >2 个 SD,证实了大头畸形的诊断。结论 CDM 应包括在妊娠晚期大头畸形的鉴别诊断中,尤其是在存在其他超声线索且母亲病史和体格检查提示 DM 的情况下。
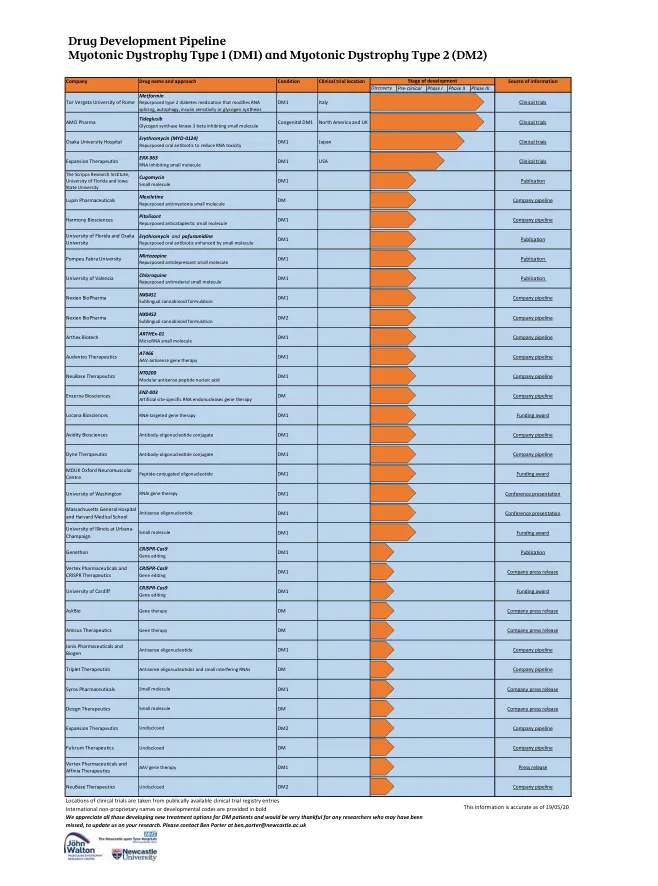
药物开发管道 1 型强直性肌营养不良症 (...
临床试验地点取自公开的临床试验注册条目 国际非专利名称或开发代码以粗体显示 我们感谢所有为糖尿病患者开发新治疗方案的人,并非常感谢任何可能被遗漏的研究人员向我们更新您的研究。请联系 Ben Porter,邮箱地址为 ben.porter@newcastle.ac.uk

Duchenne和Becker肌肉营养不良的基因检测
Duchenne肌肉营养不良DMD是最常见的肌肉营养不良,是一种严重的儿童X连锁隐性疾病,由于骨骼肌病和心肌病而导致严重的残疾。该疾病的特征是进行性,对称肌肉无力和步态障碍是由缺陷的肌营养不良蛋白基因引起的。根据2022年的系统审查和荟萃分析,DMD的全球流行率为4.8例(95%置信区间[CI],3.6至6.3),每10万人。大约三分之一的DMD案例来自从头变种,没有已知的家族史。患有DMD的婴儿通常是无症状的。表现可能早在某些患者的第一年就出现,但是临床表现最常出现在学前班期间,从2年到5年。受影响的儿童出现了步态问题,小腿肥大,正gower迹象和难以攀登楼梯。受影响的儿童运动状况可能在3到6年之间的生命状况下降,而劣化始于6至8年。大多数患者将是9至12岁的轮椅,但会保留保留的上限功能直到以后。心肌病通常发生在18岁之后。晚期并发症是心脏呼吸症(例如,由于呼吸道肌肉无力和心肌病而导致的肺功能降低)。这些严重的并发症通常是

强直性肌营养不良症 2 型中长度 CNBP 扩增等位基因...
摘要 2 型强直性肌营养不良 (DM2) 是由 CNBP 基因中的 CCTG 重复扩增引起的,该扩增包含 75 至 >11,000 个单位,具有广泛的嵌合性,因此对完全扩增的等位基因进行测序具有挑战性。为了克服这些限制,我们使用无 PCR 的 Cas9 介导纳米孔测序在 9 名 DM2 患者的单核苷酸水平上表征了 CNBP 重复扩增。使用此策略可以精确评估正常和扩增等位基因的长度,与传统方法一致,并揭示了嵌合性的程度。我们还对整个 ~50 kbp 的扩增进行了测序,这在 DM2 或任何其他重复扩增疾病中都是前所未有的。我们的方法精确地计算了重复次数并确定了短中断和不间断等位基因的重复模式。有趣的是,在扩增的等位基因中,只有两个 DM2 样本具有预期的纯 CCTG 重复模式,而其他七个样本在 3 ' 端也呈现出 TCTG 阻断,这在 DM2 患者中以前从未报道过,但在此通过正交方法得到证实。所展示的方法同时确定了重复长度、结构/基序和体细胞嵌合程度,有望改善 DM2 的分子诊断并实现更准确的基因型-表型相关性,从而在临床试验中更好地对 DM2 患者进行分层。

镰状细胞基因治疗药物事先授权表格
•必须批准适应症,年龄,并且不超过表1中列出的剂量限制。•对于所有列出的代理人,除非患者符合非偏见的PDL PA标准,否则需要在适用的情况下(如果适用)(如果适用)进行PA指示。•药物是由专门治疗DMD(即神经科医生,儿科神经科医生或物理医学和康复专家)的处方者处方或协商的。3•患者以前不得接受过levidys™或任何其他包含腺相关载体的疗法。•患者必须满足所有以下所有内容:1,6

肌发育症药物开发管道 - 2024年12月
注意:上面不包括学术机构。除了此列表外,发现阶段还有更多公司尚未公开其DM计划。单击上面的绿色箭头以获取有关第1-3阶段的临床试验信息。有关每个临床试验的位置和细节,请访问www.clinicaltrials.gov。由MDF设计,该管道正在进行开发,并基于公开可用的信息。特别感谢Nate UHL对DM研究景观的全面扫描和药物开发管道的原始愿景。有关问题,或请求对管道的更新,请通过info@myotonic.org与MDF联系。要查看肌发育症研究地图访问:www.myotonic.org/myotonic-dystrophy-research-map。

faciosculohumeral肌肉营养不良临床试验的结果度量
摘要:Facioscapulohumeral肌肉营养不良(FSHD)是一种令人衰弱的肌肉营养不良,具有变化,严重性和进展的变化。虽然仍然无法治愈这种疾病,但自从双鸡蛋ox 4(Dux4)基因的表观遗传压缩的潜在机制导致骨骼肌毒性以来,朝着FSHD疗法的进展加速了。这促进了新的疗法的快速发展,以靶向DUX4表达和下游失调,从而导致肌肉变性。这些发现和临床前翻译研究为等待临床试验评估的疗法开辟了新的途径。正如领域预期的FSHD试验一样,对于早期和II期和III期试验,需求已增长,对肌肉功能的更可靠和量化的结果度量的增长。促进纵向临床评估的高级工具将大大提高试验的潜力,以确定成功改善疾病进展或允许肌肉功能恢复的治疗剂。在这里,我们讨论了当前和新兴的FSHD结果指标以及研究人员可能在将此类措施应用于FSHD临床试验设计和实施中所面临的挑战。