XiaoMi-AI文件搜索系统
World File Search Systemexpress

IITBBS Express Vol-12日期:29.10.2024 电气科学学院(电子与通信工程) 基础科学学院(数学)Q-Exam主题&...
Shreepad Karmalkar教授处理了有关“解决问题”的会议,其中他展示了一般纪律独立策略如何帮助解决各种现实生活中的问题。一个人需要从某个领域进行专业知识来解决问题的心态是阻止人们解决问题的原因。纪律中的独立策略包括逻辑推理,反复试验,问题陈述,分析和类比的重新制定。分析涉及三个步骤:将整个部分分为部分,孤立地理解各部分,结合了对所获得的部分了解整体的理解。分析方法包括表示 /建模,划分和征服的策略以及逻辑推理。他还提到沟通,协作和批判性思维是现实生活中成功所需的三种技能。


快速行业对欧盟经济的影响
Headquartered in Oxford, England, with regional centres in London, New York, and Singapore, Oxford Economics has offices across the globe in Belfast, Boston, Cape Town, Chicago, Dubai, Frankfurt, Hong Kong, Houston, Johannesburg, Los Angeles, Melbourne, Mexico City, Milan, Paris, Philadelphia, Sydney, Tokyo, and多伦多。我们雇用400名全职员工,包括250多名专业经济学家,行业专家和业务编辑,这是最大的宏观经济学家和思想领导力专家之一。我们的全球团队在整个研究技术和思想领导能力方面都非常熟练,从计量经济学建模,场景框架和经济影响分析到市场调查,案例研究,专家小组和Web分析。

优化 PCI Express 的设计与实现...
1 研究生学者,印度班加罗尔 BIT 电子与计算机工程系 2 助理教授,印度班加罗尔 BIT 电子与计算机工程系 摘要 PCIe(外围组件互连快捷)协议对于在计算机外围设备(如显卡和网卡等)之间建立高速数据通信至关重要。此通信协议以数据包格式传输数据,每个数据包包含数据和目标地址以及其他准确传输数据的基本信息。本文重点研究物理层通过降低延迟参数实现高速数据传输。为了最大限度地减少干扰并提高可靠性,物理层使用加扰技术,并使用 8b – 10b 编码技术进行同步和错误检测。此外,SIPO 和 PISO 转换数据格式以提高效率和准确性。该设计使用针对 45nm 工艺技术的 Cadence 编译器实现。该设计具有延迟效率,路径延迟为 5.0ns,工作频率为 200MHz,功耗为 1.2mw,面积为 1999µm²。关键词:PCIe、加扰器和解扰器、8b-10b 编码器和 10b-8b 解码器、数据包、PISO 和 SIPO。I. 简介 PCIe(外围组件互连快速)是一种用于计算机的高速串行扩展总线,它取代了目前计算机中未使用的并行总线,如 PCI-PCI-X 和 AGP。因此,它具有速度、可扩展性和性能改进等特点 PCIe 基于点对点连接,其中每个设备直接连接到终端节点,最常见的是 CPU 或芯片组。这消除了共享总线设计带来的争用,从而提高了整个系统的性能,并降低了延迟。点对点连接大多基于数据包,服务质量 (QoS) 可实现低延迟。这些功能对于需要加载和传输大量数据的任何应用程序都至关重要,例如游戏、计算密集型应用程序 HPC 或商业智能和分析应用程序。借助 PCIe 接口中使用的热插拔、错误检测和纠正 (EDC) 功能,可以提高系统可靠性。PCIe 是一种串行、点对点和基于数据包的协议,由 PCI-SIG(PCI 特别兴趣小组)维护和定义。PCIe 实际上是 PCI 的替代品,PCI 是一种芯片并行总线协议。PCIe 是一种高速串行;这是选择 PCIe 来支持此机器高速数据传输的主要原因,用于连接的计算机扩展总线标准

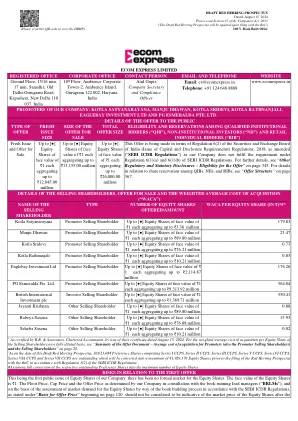
Ecom Express Limited注册办公室...
*由B.B.&Associates,特许会计师,通过2024年8月15日的证书。以完全稀释的基础上的销售股东平等份额的加权平均收购成本,请参阅“要约文件的摘要 - 促进者的平均收购成本(也是推动者出售股东)和销售股东”和第28页。^截至此红鲱鱼招股说明书的日期,有10,623,088份优先股包括I系列CCP,II系列CCP,III系列CCPS,系列VI CCPS,VI CCPS系列VI CCPS,通过CCPS和VII CCPS系列串联VII CCPS系列均可将其最高转化为81,92929,370 Eqection forder formation formation forkes forkects forkestion forkes forthers forkes force fors fore fors fors fore fors fore fors fore for 81,92,370 equeS组成的列表。根据SEBI ICDR法规的第5(2)条,ROC。#shessum将各个未偿还股份的全部转换为最大股票数量。

航空货运和快递包裹 - Atrax Group
侧面护栏和多个上摆式钢制挡块可用于容纳各种尺寸的集装箱,并有助于放置货物。任何甲板都可以是单秤平台或分体式双甲板,每半甲板都内置有单独的称重传感器。每半甲板都可以独立使用,允许同时称量两个单独的集装箱,或者将整个甲板模块用作大型 ULD 的组合秤。双甲板单元必须与 Atrax CDI-1600 或 920i 双通道数字重量指示器连接,以显示单个甲板重量和总重量。所有其他单甲板都可以与任何 Atrax 数字重量指示器 (DWI) 连接。

通过 AI Express 将数据转化为智能 - 万事达卡
Brighterion AI 将使用您的历史数据构建您的个性化模型,以确保立即节省开支。这些数据和未来的数据来自任何来源,格式也各不相同,Brighterion AI 使用人工智能和机器学习来丰富这些数据。无监督和监督学习会随着时间的推移改善结果,创建随着欺诈者不断变化的行为而成长和成熟的 AI 模型。

向 Patriot Express 宠物主人发出的警告信息
2022 年 7 月 15 日 — 认真对待您的宠物。作为有五次海外旅行经历的宠物主人,我深知这些重要事项的风险、费用和极度担忧……
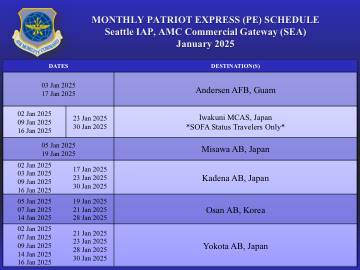
每月爱国者快车 (PE) 时刻表西雅图......
2025 年 1 月 2 日 2025 年 1 月 7 日 2025 年 1 月 9 日 2025 年 1 月 14 日 2025 年 1 月 16 日