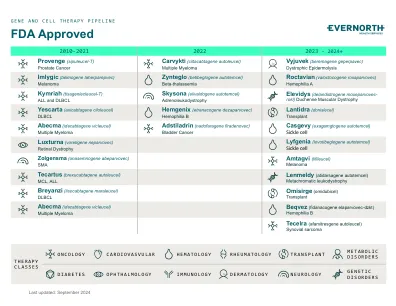
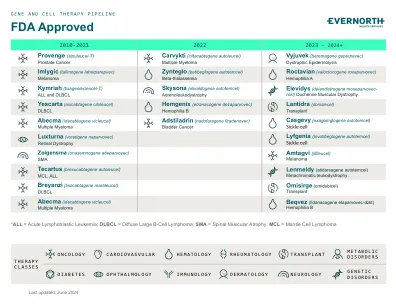
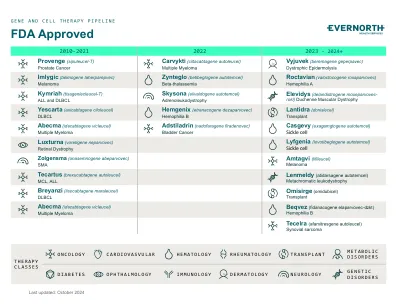
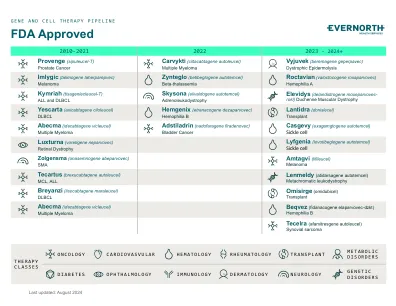
XiaoMi-AI文件搜索系统
World File Search Systemfda

fda
FDA Jacqueline Corrigan-Curay,J.D.,M.D。药物评估与研究中心校长中心主任JACQUELIN CORRIGAN-CURAY,J.D.,M.D.是FDA药物评估与研究中心(CDER)的首席副副主任(CDER),她在战略性计划中为CDER提供了促进CDER的行政领导,以促进CDER提供安全,有效,高品质的美国公众。在担任这一角色之前,Corrigan-Curay博士是CDER医疗政策办公室的主任,领导了医疗政策计划的开发,协调和实施,包括现实证据,包括在药物开发和处方药物促进中使用技术。在加入FDA之前,她曾在国立卫生研究院(NIH)的国家心脏,肺和血液研究所(NHLBI)的董事直接办公室担任高级医疗官。她还曾担任NIH科学政策办公室生物技术活动办公室主任。Corrigan-Curay博士从哈佛大学法学院获得了法律学位,马里兰大学医学院的医学学位以及马萨诸塞州剑桥的哈佛/拉德克利夫学院的科学史学士学位。Celia Witten,博士,M.D。 生物制剂评估与研究中心副主任Celia M. Witten博士,M。D。是食品和药物管理局(FDA/CBER)生物制剂评估与研究中心副主任。 在2005年至2016年之间,她曾在FDA/CBER担任蜂窝,组织和基因治疗办公室主任。 她的教育背景包括学士学位。Celia Witten,博士,M.D。生物制剂评估与研究中心副主任Celia M. Witten博士,M。D。是食品和药物管理局(FDA/CBER)生物制剂评估与研究中心副主任。 在2005年至2016年之间,她曾在FDA/CBER担任蜂窝,组织和基因治疗办公室主任。 她的教育背景包括学士学位。生物制剂评估与研究中心副主任Celia M. Witten博士,M。D。是食品和药物管理局(FDA/CBER)生物制剂评估与研究中心副主任。在2005年至2016年之间,她曾在FDA/CBER担任蜂窝,组织和基因治疗办公室主任。她的教育背景包括学士学位。在1996年至2005年之间,她曾在设备和放射健康中心(CDRH)的设备评估办公室担任一般,修复和神经系统设备的主任。在FDA之前,她在华盛顿特区的国家康复医院(NRH)担任执业医师超过10年。在普林斯顿大学(Magna Cum Laude)获得博士学位。来自斯坦福大学和医学博士来自迈阿密大学医学院。除了她的学术成就外,她还获得了物理医学和康复的董事会认证。Sandra Retzky,D.O.,J.D.,M.P.H. 专员桑德拉·桑迪·雷茨基(Sandra“ Sandy” Retzky)办公室的孤儿产品开发办公室主任是FDA孤儿产品开发办公室(OOPD)的主任。 Sandy于2016年加入了该机构,并在烟草产品中心担任医学审查员,曾在烟草产品的营销机构中申请。 在2019年,桑迪(Sandy)成为了CBER医学审查员,并花了几年的时间管理许多基因和细胞疗法文件来治疗罕见疾病。 桑迪最初接受过药剂师的培训,并从伊利诺伊大学药学院获得学位。 她毕业于芝加哥的一所骨病医学院中西部大学。 桑迪的医疗证书包括妇产科和妇科董事会认证,泌尿妇女学研究奖学金培训以及在特拉华州执业的许可。Sandra Retzky,D.O.,J.D.,M.P.H.专员桑德拉·桑迪·雷茨基(Sandra“ Sandy” Retzky)办公室的孤儿产品开发办公室主任是FDA孤儿产品开发办公室(OOPD)的主任。Sandy于2016年加入了该机构,并在烟草产品中心担任医学审查员,曾在烟草产品的营销机构中申请。在2019年,桑迪(Sandy)成为了CBER医学审查员,并花了几年的时间管理许多基因和细胞疗法文件来治疗罕见疾病。桑迪最初接受过药剂师的培训,并从伊利诺伊大学药学院获得学位。她毕业于芝加哥的一所骨病医学院中西部大学。桑迪的医疗证书包括妇产科和妇科董事会认证,泌尿妇女学研究奖学金培训以及在特拉华州执业的许可。在从事医学多年后,桑迪从宾夕法尼亚大学的沃顿大学获得了MBA学位,并在制药和生物技术行业工作