XiaoMi-AI文件搜索系统
World File Search Systemgrnas
![植物生物技术 23.0611a (2023) [adv.pub.]](/simg/8\886d12a05c6e0ea5545c8dfe272b81f8a9782723.webp)
植物生物技术 23.0611a (2023) [adv.pub.]
摘要 马铃薯 (Solanum tuberosum L.) 具有四倍体基因组。要制造缺乏特定基因功能的突变体,必须将突变引入所有四个基因等位基因中。为了实现这一目标,我们开发了一种强大的基因组编辑工具 CRISPR/dMac3-Cas9,它安装了翻译增强子 dMac3,大大增加了下游开放阅读框的翻译。采用三种向导 RNA (gRNA) 的 CRISPR/dMac3-Cas9 系统大大提高了突变的发生率。该系统能够创建颗粒结合淀粉合酶 (GBSS) 和淀粉分支酶 (SBE) 的 4 等位基因突变体。这些突变体显示出功能缺陷的特征,表明我们成功地对马铃薯四倍体基因组进行了有效的基因组编辑。在这里,我们展示了使用 GBSS1 基因突变体时 gRNA 数量对目标基因有效诱变的影响。采用三个 gRNA 基因的 CRISPR/dMac3-Cas9 比采用两个 gRNA 的 CRISPR/dMac3-Cas9 实现了更高的突变效率,表明突变效率受靶区域 gRNA 数量的剂量效应影响。SBE3 基因的等位基因含有导致 gRNA 序列差异的 SNP,但这些 gRNA 有效发挥作用。然而,诱导了许多重排事件和大量缺失。这些结果支持 gRNA 与靶序列准确结合的重要性,这可能为避免脱靶位点的意外突变提供线索。

多重、单细胞 CRISPRa 筛选细胞类型特异性调控元件
基于 CRISPR 的基因激活 (CRISPRa) 是一种通过以组织/细胞类型特异性的方式靶向启动子或增强子来上调基因表达的策略。在这里,我们描述了一个实验框架,该框架将高度多路复用的扰动与单细胞 RNA 测序 (sc-RNA-seq) 相结合,以识别细胞类型特异性、CRISPRa 响应的顺式调控元件及其调控的基因。将许多 gRNA 的随机组合引入许多细胞中的每一个,然后对其进行分析并分成测试组和对照组,以测试 CRISPRa 对增强子和启动子的扰动对邻近基因表达的影响。将该方法应用于 493 个 gRNA 文库,这些 gRNA 靶向 K562 细胞和 iPSC 衍生的兴奋性神经元中的候选顺式调控元件,我们识别出能够特异性上调预期靶基因且 1 Mb 内没有其他邻近基因的 gRNA,包括导致神经元中六种自闭症谱系障碍 (ASD) 和神经发育障碍 (NDD) 风险基因上调的 gRNA。一致的模式是,单个增强子对 CRISPRa 的响应受细胞类型的限制,这意味着成功激活基因依赖于染色质景观和/或其他反式因子。本文概述的方法可能有助于大规模筛选以细胞类型特异性方式激活基因的 gRNA。

使用 Benchling 教程设计 gRNA:Myriam Sévigny 撰写,2022 年基因组生物学单元由 HiLIFE 和赫尔大学医学院支持
• 对于基因敲除,gRNA 通常靶向 5' 组成性表达的外显子,这降低了由于可变剪接而从 mRNA 中移除目标区域的可能性。 • 根据经验,避免靶向编码蛋白质 N' 末端附近氨基酸的位点,以减轻细胞使用注释起始密码子下游的替代 ATG 的能力。同样,避免靶向编码蛋白质 C' 末端附近氨基酸的位点,以最大限度地增加产生无功能等位基因的机会。对于 1 千碱基基因,由于潜在靶向位点每 8 个核苷酸中出现约 1 个,将 gRNA 限制在蛋白质编码区域的 5 – 65% 仍将导致有数十种 gRNA 可供选择。有这么多可能性,选择具有优化序列的 gRNA 是首要任务。 • 如果可能,设计 gRNA 以靶向编码已知必需蛋白质结构域的外显子。这种方法的好处是,即使非移码等位基因在重要的蛋白质结构域中出现时也可能改变蛋白质的功能。

1 用于多拷贝基因整合的着陆垫系统...
图 1. SD108 中全基因组整合位点的计算机筛选算法。(A)选择基因间位点中的 gRNA 进行 iCas9 介导的整合。扫描基因组中的“NGG”PAM 以获得向导 RNA 文库。筛选 gRNA 以尽量减少潜在的脱靶,并根据其基因组位置进行过滤。(B)结合各种因素对实验筛选的基因组位点进行优先排序。根据寡核苷酸合成和质粒克隆标准对 gRNA 及其相应的同源臂进行改进。实施设计规则以确保菌株稳定性,避免破坏调控元件并包括基因必需性信息,同时添加基因密度作为开放染色质的代理。结合转录组学数据来选择靠近转录活性基因的位点。

Issatchenkia Orientalis
图1。SD108中全基因组整合位点的硅筛选算法算法。 (a)用于ICAS9介导的整合的基因基因座中的GRNA。 扫描基因组以获取“ NGG” PAM以获得指南RNA库。 筛选GRNA以最大程度地减少潜在的脱靶,并根据其基因组位置过滤。 (b)纳入各种因素以优先考虑基因组基因局进行实验筛查。 GRNA及其相应的同源臂是根据寡核苷酸合成和质粒克隆标准来完善的。 设计规则是通过避免调节元素的破坏和包括基因本质信息的中断来确保应变稳定性的,而基因密度则是添加基因密度作为开放染色质的代理。 转录组数据纳入了接近转录活性基因的选择位置。算法。(a)用于ICAS9介导的整合的基因基因座中的GRNA。扫描基因组以获取“ NGG” PAM以获得指南RNA库。筛选GRNA以最大程度地减少潜在的脱靶,并根据其基因组位置过滤。(b)纳入各种因素以优先考虑基因组基因局进行实验筛查。GRNA及其相应的同源臂是根据寡核苷酸合成和质粒克隆标准来完善的。设计规则是通过避免调节元素的破坏和包括基因本质信息的中断来确保应变稳定性的,而基因密度则是添加基因密度作为开放染色质的代理。转录组数据纳入了接近转录活性基因的选择位置。

基于 CRISPR 的配对引导 RNA 抑制 VEGF 治疗脉络膜新生血管
在临床前研究中,利用单个 gRNA 对血管内皮生长因子 A (Vegfa) 进行基于成簇的规律间隔短回文重复序列 (CRISPR) 的基因组破坏可抑制脉络膜新生血管 (CNV),为新生血管性年龄相关性黄斑变性 (AMD) 的长期抗血管生成治疗提供了前景。使用 CRISPR-CRISPR 相关核酸内切酶 (Cas9) 和多个向导 RNA (gRNA) 进行基因组编辑可以通过用基因截断增强插入-缺失 (indel) 突变来增强基因消融效果,但也可能增加脱靶效应的风险。在本研究中,我们比较了腺相关病毒 (AAV) 介导的 CRISPR-Cas9 系统使用单个和配对 gRNA 靶向 Vegfa 基因中在人类、恒河猴和小鼠中保守的两个不同位点的有效性。配对 gRNA 在体外增加了人类细胞中 Vegfa 基因消融率,但在体内并未增强小鼠眼中的 VEGF 抑制。与单个 gRNA 系统相比,使用配对 gRNA 的基因组编辑也显示出相似程度的 CNV 抑制。使用通过测序 (GUIDE-seq) 实现的全基因组无偏双链断裂 (DSB) 识别进行的无偏全基因组分析揭示了由第二个 gRNA 引起的微弱脱靶活性。这些发现表明,使用两个 gRNA 进行体内 CRISPR-Cas9 基因组编辑可能会增加基因消融,但也可能会增加脱靶突变的潜在风险,而针对 Vegfa 基因中的另一个位点作为新生血管性视网膜疾病治疗的功能益处尚不清楚。

TDG CAS9-CKO策略
过程如下:在体外转录GRNA。cas9和grnas微注射到C57BL/6JGPT小鼠的受精卵中。受精卵被移植以获得通过PCR和靶标扩增子测序确认的阳性F0小鼠。通过与C57BL/6JGPT小鼠配对阳性F0产生小鼠获得稳定的F1生成小鼠菌株,并通过PCR和靶向Agplicon测序对所需的突变等位基因进行确认。

在大豆中的CRISPR质量表达,用于简化planta
摘要 - 这项工作的目的是开发一种在大豆(Glycine Max)胚胎中创建和验证CRISPR-CAS系统和不同GRNA的方法。两个模型基因用于用一个GRNA或部分基因缺失的简单突变。通过使用经典限制酶克隆方法将启动子 + grna2插入CRISPR转换向量中,或通过将启动子 + GRNA2取代和插入启动子 + GRNA2。向量成功地构造了一个和两个grnas。大豆中的农杆菌介导的瞬时转化是为了测试GRNA和系统本身的质量(表达盒)。通过酶消化后DNA富集后转化的胚胎中检测到了简单的突变和基因缺失,然后是聚合酶链反应和测序,这表明CRISPR-CAS系统和指南在起作用。该方案可用于加速基于CRISPR的基因组编辑策略,用于大豆的遗传转化。

合成 CRISPR/Cas9 试剂促进基因组编辑...
CRISPR/Cas9 已成为斑马鱼基因组编辑的有力工具,它允许使用 DNA 模板和同源定向修复 (HDR) 快速产生功能丧失突变和特定等位基因的敲入。我们检查了合成的、化学修饰的 gRNA 的效率,并证明与重组 Cas9 蛋白结合可诱导插入缺失和大型基因组缺失。我们开发了一种体内遗传检测方法来测量 HDR 效率,并利用该检测方法来测试改变模板设计对 HDR 的影响。利用合成的 gRNA 和线性 dsDNA 模板,我们成功地在多个基因组位点进行了荧光团的敲入,并证明了以高效率通过种系传递。我们证明合成的 HDR 模板可用于敲入细菌硝基还原酶 (ntr),以促进特定细胞类型的谱系消融。总的来说,我们的数据证明了结合合成 gRNA 和 dsDNA 模板在体内进行同源定向修复和基因组编辑的实用性。
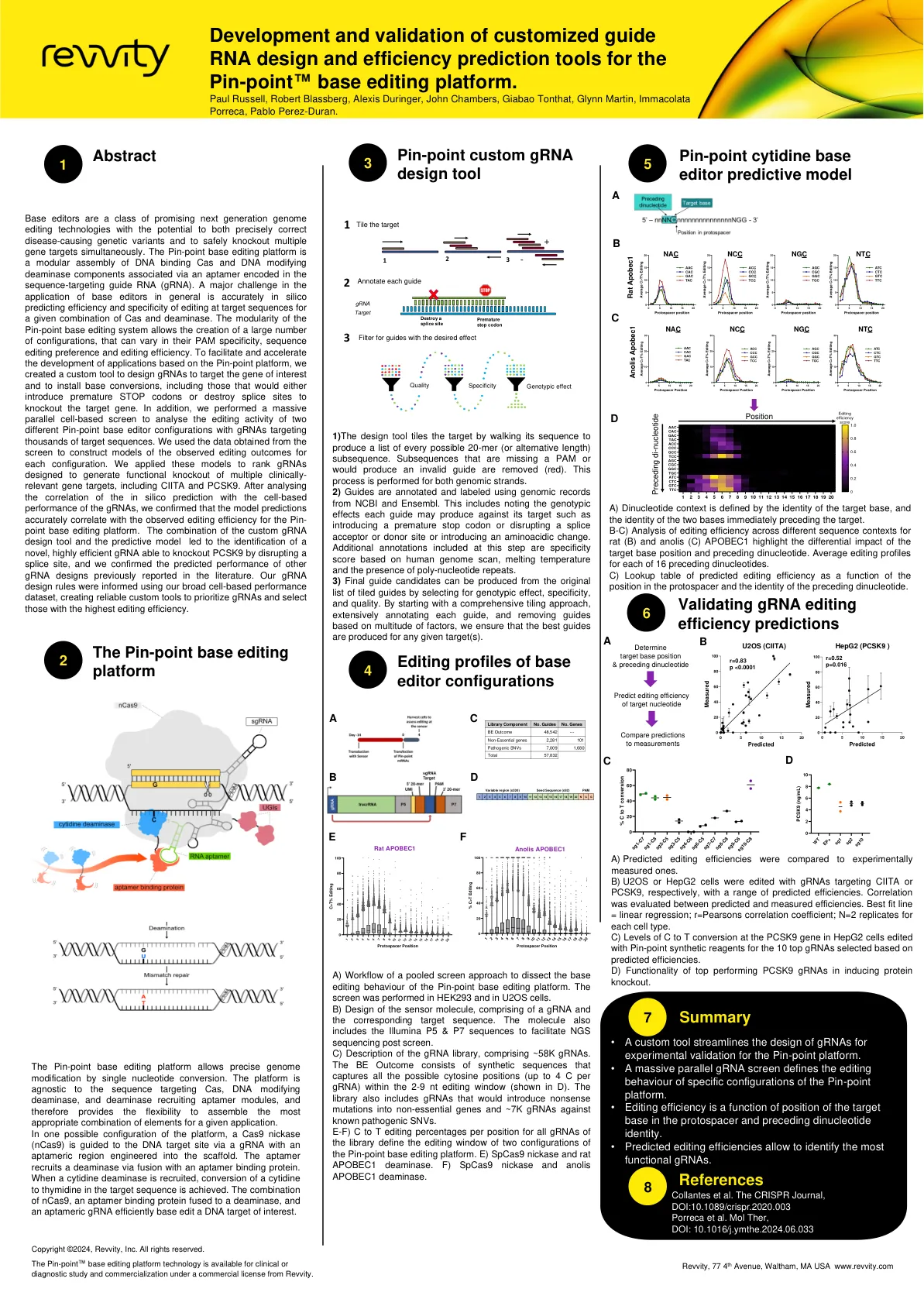
精准定制 gRNA 设计工具
碱基编辑器是一类有前途的下一代基因组编辑技术,既可以精确纠正致病的遗传变异,又可以同时安全地敲除多个基因靶标。Pin-point 碱基编辑平台是一个模块化组装的 DNA 结合 Cas 和 DNA 修饰脱氨酶组件,它们通过序列靶向向导 RNA (gRNA) 中编码的适体连接。碱基编辑器应用中的一个主要挑战是准确地通过计算机预测给定 Cas 和脱氨酶组合在目标序列上的编辑效率和特异性。Pin-point 碱基编辑系统的模块化允许创建大量配置,这些配置的 PAM 特异性、序列编辑偏好和编辑效率可能有所不同。为了促进和加速基于 Pin-point 平台的应用程序开发,我们创建了一个自定义工具来设计 gRNA 以靶向感兴趣的基因并安装碱基转换,包括那些会引入过早的终止密码子或破坏剪接位点以敲除目标基因的转换。此外,我们进行了大规模并行细胞筛选,以分析两种不同的 Pin-point 碱基编辑器配置的编辑活动,其中 gRNA 靶向数千个目标序列。我们使用从筛选中获得的数据构建了每种配置观察到的编辑结果的模型。我们应用这些模型对设计用于产生多个临床相关基因靶标(包括 CIITA 和 PCSK9)功能性敲除的 gRNA 进行排序。在分析了计算机预测与 gRNA 的细胞性能之间的相关性后,我们确认模型预测与 Pin-point 碱基编辑平台观察到的编辑效率准确相关。自定义 gRNA 设计工具和预测模型的结合导致识别出一种新型、高效的 gRNA,它能够通过破坏剪接位点来敲除 PCSK9,并且我们确认了文献中先前报道的其他 gRNA 设计的预测性能。我们的 gRNA 设计规则是使用我们广泛的基于细胞的性能数据集得出的,从而创建了可靠的自定义工具来优先考虑 gRNA 并选择具有最高编辑效率的 gRNA。