XiaoMi-AI文件搜索系统
World File Search Systemgrnas
![植物生物技术 37: 20.0404b (2020) [adv.pub.]](/simg/7\79f64403bd75f8328112fa08a0b4888c68c1ea25.webp)
植物生物技术 37: 20.0404b (2020) [adv.pub.]
摘要 利用 CRISPR/Cas9 进行基因组编辑对普通小麦非常有用,因为普通小麦具有异源六倍体的特性,并且可以同时在三个同源基因中诱发突变。虽然农杆菌介导的转化在基因组编辑方面具有优势,但它在小麦中仍然效率低下且需要相对较长的时间。因此,使用具有高效体内诱变功能的向导 RNA (gRNA) 是在短时间内产生基因组编辑突变系的关键因素之一。在本研究中,我们针对普通小麦中的三个基因,建立了一种快速检测由基因枪瞬时表达系统诱导的突变的方法。在未成熟的小麦胚中实现了 gRNA 和 Cas9 的基因枪瞬时表达。一周后使用 PCR-RFLP 检测到突变,并通过基因组克隆测序进行验证。我们确认了几种类型的突变,这些突变的发生率取决于靶序列。此外,在农杆菌转化的植物中,以较高速率编辑的靶标处的突变频率往往更高。这些结果表明,这种快速检测编辑突变的方法可用于多种应用,例如筛选目标序列或修饰载体以实现小麦中有效的 CRISPR/Cas9 基因组编辑。

通过将CRISPR/CAS9系统电穿孔到Zygotes的一步生成多个基因编辑的猪,以减少异种抗原生物合成
摘要:异种抗原引起过度急性排斥并限制了种间异种移植的成功。因此,参与异种抗原生物合成的基因,例如GGTA1,CMAH和B4GALNT2,是改善异种移植结果的关键靶标。在这项研究中,我们引入了一种CRISPR/CAS9系统,同时使用电穿孔来靶向GGTA1,CMAH和B4GALNT2,以一步一代生成多个基因编辑的猪而没有异种抗原。首先,我们优化了针对GGTA1和CMAH的引导RNA(GRNA)相对于基因编辑的效率,并将电穿孔的胚胎与优化的GRNA和CAS9转移到受体Gilts中。接下来,当GGTA1,CMAH和B4GALNT2同时靶向GGTA1,CMAH和B4GALNT2时,我们优化了CAS9蛋白的浓度,并使用优化的条件同时靶向基因。我们实现了GGTA1 / CMAH双重编辑的猪和GGTA1 / CMAH / B4GALNT2三重编辑的猪的一步生成。免疫组织学分析表明异种抗原的下调。然而,这些多个基因编辑的猪是遗传镶嵌物,未能敲除一些异种抗原。尽管应解决镶嵌性,但电穿孔技术可能会成为旨在改善猪至人类异种移植的猪中一步生成多个基因修饰的主要方法。

使用CRISPR/CAS9系统在DMD基因中淘汰外显子48
抽象背景:DMD基因中的框架突变导致Duchenne肌肉营养不良(DMD),这是一种神经肌肉进行性遗传疾病。在DMD患者中,缺乏肌营养不良蛋白会导致进行性肌肉变性,从而导致心脏和呼吸道衰竭导致过早死亡。目前,尚无对DMD的某些治疗方法。dmd基因是2.2巨型碱基对的人类基因组中最大的基因,并包含79个外显子。在过去的几年中,基因疗法被认为是一种有希望的DMD治疗,在各种基因编辑技术中,CRISPR/CAS9系统被证明更加精确和可靠。这项研究的目的是评估使用一对SGRNA敲除外显子48的可能性。方法:一对指导RNA(GRNA)设计用于切割DMD基因,并诱导外显子48的缺失。将GRNA转接为HEK-293细胞系,然后通过PCR分析基因组DNA中的DELENEC,然后通过PCR和随后的Sanger测序分析。结果:外显子48被成功删除,因此外显子47连接到外显子49。结论:此结果表明CRISPR/CAS9系统可用于精确编辑DMD基因。关键字:CRISPR/CAS9,肌营养不良,基因编辑,肌肉营养不良简介
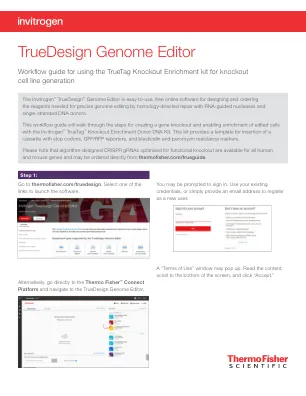
TrueDesign 基因组编辑器
对于使用 CRISPR 技术的敲除富集实验,设计步骤中选择的 gRNA 和 Invitrogen™ TrueTag™ 供体引物将自动包含在内。该工具还将添加 TrueTag Knockout Enrichment Donor DNA Kit,其中包含所需的供体模板、PCR 试剂和清理试剂盒,以生成可用于转染的供体 DNA。还包括 Invitrogen™ TrueTag™ 验证引物,用于进行连接分析,以确保供体模板正确插入。

全面的 Bioconductor 生态系统,用于跨核酸酶和技术设计 CRISPR 引导 RNA
CRISPR 介导的基因扰动研究的成功高度依赖于 gRNA 的质量,并且已经开发了几种工具来实现最佳的 gRNA 设计。然而,这些工具并不都适用于最新的 CRISPR 模式或核酸酶,也没有提供全面的注释方法或用于高级 CRISPR 应用的可扩展性。在这里,我们介绍了一个新的 R 包生态系统,它能够为多种 CRISPR 技术实现高效的 gRNA 设计和注释,包括 CRISPR 敲除、CRISPR 激活 CRISPR 干扰和 CRISPR 碱基编辑。核心包 crisprDesign 提供了一个全面、用户友好且统一的界面,可通过几种比对方法添加靶向和脱靶注释、丰富的基因和 SNP 注释以及十几个靶向和脱靶活动分数。这些功能适用于任何 RNA 或 DNA 靶向核酸酶,包括 Cas9、Cas12 和 Cas13。我们通过为三个案例研究设计最佳 gRNA 来说明我们工具的普遍适用性:使用碱基编辑器 BE4max 平铺 BRCA1 的 CRISPRbe 库、使用 CasRx 平铺 CD46 和 CD55 的 RNA 靶向库以及使用 CRISPRa 激活 MMP7。我们的 R 软件包套件是开源的,并通过 Bioconductor 项目部署,以方便 CRISPR 社区使用它们。

多重引导RNA表达系统及其对植物基因组工程的功效
摘要背景:化脓性链球菌 CRISPR 系统由 Cas9 内切酶 (Sp Cas9) 和含有靶标特异性序列的单链向导 RNA (gRNA) 组成。理论上,Sp Cas9 蛋白可以切割与基因组中结合的 gRNA 一样多的靶标位点。结果:我们引入了一种无 PCR 的多 gRNA 克隆系统来编辑植物基因组。该方法包括两个步骤:(1)在 pGRNA 载体中的 tRNA 和 gRNA 支架序列之间克隆两个单链寡核苷酸片段的退火产物,该片段的每条链上都含有互补的靶标结合序列;(2)使用 Golden Gate 组装方法将来自几个 pGRNA 载体的 tRNA-gRNA 单元与含有 Sp Cas9 表达盒的植物二元载体组装在一起。我们通过进行靶向深度测序验证了多重 gRNA 表达系统在野生烟草(Nicotiana attenuata)原生质体和转化植物中的编辑效率和模式。Sp Cas9-gRNA 的两次近端切割大大提高了编辑效率,并在两个切割位点之间诱导了较大的缺失。结论:这种多重 gRNA 表达系统能够高通量生产单个二元载体,并提高植物基因组编辑的效率。关键词:CRISPR-Cas9、金门组装、多重 gRNA、植物基因组编辑

multicrispr:用于主要编辑和数千个目标的并行定位的 gRNA 设计
近年来,通过 Crispr/Cas9 技术靶向编码基因组引入单核苷酸缺失/插入已成为一种标准程序。它迅速催生了多种方法,例如 Prime Editing、Crispr/Cas9 辅助 APEX 邻近标记蛋白质或同源定向修复 (HDR),但支持这些方法的生物信息学工具却落后了。新应用通常需要特定的向导 RNA (gRNA) 设计功能,而通用的 gRNA 设计工具却严重缺失。在这里,我们回顾了 gRNA 设计软件并介绍了 multicrispr,这是一种基于 R 的工具,旨在设计单个 gRNA 以及并行靶向许多基因组位点的 gRNA 库。该软件包易于使用,可检测、评分和过滤 gRNA 的效率和特异性,可视化和汇总每个目标或 Crispr/Cas9 序列的结果,最后返回基因组范围以及首选的、无脱靶 gRNA 序列。为了通用,multicrispr 定义并实施了一个基因组算法框架,作为轻松适应尚未出现的技术的基础。其性能和新的 gRNA 设计概念(例如针对 gRNA 库的目标集特定过滤)使 multicrispr 成为处理类似筛选方法时的首选工具。

GRNA设计:其进化如何影响CRISPR/ ... div>
摘要:在过去的十年中,基于基于RNA引导的核酸酶的相对较新的基因编辑工具的出现,基因工程进行了革命:CRISPR/CAS9系统。自1987年的第一项报告和2007年作为细菌防御机制的表征以来,该系统引起了极大的兴趣和研究的关注。CRISPR系统可为细菌免疫免受入侵遗传物质的影响。但是,通过序列和结构的特定修改,它成为一个精确的编辑系统,能够修改各种生物的基因组。这些修饰的重新确定包括各种方法,包括开发更准确的核酸酶,对细胞环境和表观遗传条件的理解以及重新设计的指南RNA(GRNA)。考虑到CRISPR/CAS9系统正确性能的重要性,我们的范围将强调后一种方法。因此,我们介绍了过去和最新指南基于RNA Web的设计工具的概述,突出了多年来其计算架构和GRNA特征的演变。我们的研究解释了使用机器学习技术,神经网络和GRNA/目标相互作用数据来启用预测和分类的计算方法。本评论可以打开一个动态社区的大门,该社区使用最新的算法来优化和创建有前途的GRNA,适合现代CRISPR/CAS9工程。

CRISPR/Cas12a (Cpf1) 引导RNA序列的鉴定
CRISPR(成簇的规律间隔的短回文重复序列)或 CRISPR 相关(Cas)系统已成为一种主要的基因编辑工具。使用 CRISPR 进行基因编辑需要 Cas 蛋白和相应的向导 RNA(gRNA)。然而,低切割效率和脱靶效应会阻碍 CRISPR/Cas 系统的应用。因此,确定特定的 gRNA 至关重要。在生物传感器应用中,由于 Cas12a(Cpf1)的反式切割活性,CRISPR/Cas12a 可以增强识别靶基因的特异性和灵敏度。mtDNA D 环序列是 mtDNA 中最易变的部分,使其适合区分物种。因此,本研究的目的是通过计算机模拟确定野猪 mtDNA D 环的 gRNA 序列。在 GenBank 数据库的帮助下,使用 Benchling 应用程序预测候选 gRNA。随后,使用 BLAST 核苷酸对 gRNA 候选物进行同源性差异分析,并使用 Jalview 进行错配测试。在几个候选物中,候选物 1 被选为最佳选择,脱靶值为 99.8。与竞争对手的同源性差异分析和与 Sus 属的错配测试分别产生了较高的 E 值和较高的百分比值。这表明候选物不会识别其他物种,但可以检测 Sus scrofa 物种的成员。这些 gRNA 候选物可以选择性地且灵敏地应用于生物传感器,以检测肉类掺假。

在番茄中应用CRISPR/CAS9介导的基因编辑
crispr-/cas9介导的基因编辑已在包括番茄在内的许多食品作物中证明。番茄(Solanum lycopersicum)既是重要的粮食作物,又是一种模型植物物种,已广泛用于研究基因功能,尤其是与水果生物学有关的植物。这种双重性在目的中与随时可用的资源(突变种群,基因组序列,转化方法)相结合,使番茄成为基因编辑的理想候选者。我们实验室通常使用的CRISPR/CAS9系统已应用于各种番茄基因型和野生物种solanum pimpinellium。矢量系统基于金门克隆技术。盒,该基因既赋予对卡纳米霉素的抗性,Kanamycin是由CAMV 35S启动子驱动的人类密码子驱动的Cas9,并在控制拟南芥U6 Polymerase促进剂的控制下引导RNA(GRNA)是组装成T-Dna的casss cass9。通常,我们设计了每个基因靶标的两个GRNA的CRISPR/CAS9构建体。但是,我们已经成功地包括多达八个grnas,以同时针对多个基因和区域。将CRISPR-/CAS9设计的构建体引入番茄中是通过基于基于NPTII基因的存在的培养基的培养基的培养基感染的转化方法来实现的。本章详细介绍了CRISPR/CAS9构建体和基因型分析(基于PCR的扩增子测序和T7核酸内切酶)的方法。