XiaoMi-AI文件搜索系统
World File Search Systemhp

期刊 - HP 实验室
顾问 Frank Harry W. Brown,集成电路业务部,加利福尼亚州圣克拉拉 » Frank J. Calvillo,格里利存储部。科罗拉多州格里利* Harry Chou,微波系统部,加利福尼亚州圣霍萨 Derek I Dang,系统支持部,加利福尼亚州山景城» Rajesh Desai,商业系统部,加利福尼亚州库比蒂诺 • Kevin G. Ewert,集成系统部,加利福尼亚州桑尼维尔 • Bernhard Fischer,伯布林根医疗部,德国伯布林根» Douglas Gennetten,格里利硬拷贝部,科罗拉多州格里利» Gary Gordon,惠普实验室,加利福尼亚州帕洛阿尔托» Matt J. Marline,系统技术部,加利福尼亚州罗斯维尔 • Bryan Hoog,Lake Stevens 仪器部,华盛顿州埃弗雷特» Grace Judy,格勒诺布尔网络部,加利福尼亚州库比蒂诺» Roger L. Jungerman,• 技术部,圣霍萨。加利福尼亚州 • Paula H. Kanarek,喷墨组件部门,科瓦利斯。俄勒冈州 • Thomas F Kraemer,科罗拉多斯普林斯部门。科罗拉多州科罗拉多斯普林斯» Ruby B. Lee,网络系统集团。加利福尼亚州库比蒂诺 • Bill Lloyd,日本惠普实验室,日本川崎» Alfred VXI Waldbronn 分析部门。德国瓦尔德布龙» Michael P. Moore,科罗拉多州洛夫兰 VXI 系统部门» Shelley I. Moore,加利福尼亚州圣地亚哥打印机部门,William Software 部门» Dona L. Merrill,全球客户支持部门。加利福尼亚州山景城* William M. Mowson,开放系统软件部,马萨诸塞州奥索里尼 » Steven J. Narciso,VXI 系统部,科罗拉多州洛夫兰 » Garry Orsolini,软件技术部,罗斯维尔。加利福尼亚州 • Raj Oza,外设技术部,山景城。加利福尼亚州 » Han Tian Phua,亚洲外设部,新加坡 » Ken Poulton,惠普实验室,加利福尼亚州帕洛阿尔托 系统 Fort Riebesell,博布林根仪器部,博布林根。德国» Marc Sabaiella,软件工程系统部,科罗拉多州柯林斯堡 • Michael B. Bristol,集成电路业务部,俄勒冈州科瓦利斯» Philip Stenton,惠普布里斯托尔实验室,英国布里斯托尔» Beng-Hang Tay,新加坡网络运营部,新加坡» Stephen R. Undy,系统技术部,科罗拉多州柯林斯堡 • Richard B.Wells,磁盘内存部,爱达荷州博伊西 • Jim Wiilits,部门。和系统管理部,科罗拉多州柯林斯堡 » Koichi Yanagawa。神户仪器部。日本神户 » Dennis C. York,科瓦利斯分部。科瓦利斯。俄勒冈州» Barbara Zimmer,企业工程部,加利福尼亚州帕洛阿尔托

HP笔记本电脑17-CP3005DX
(1A)并非所有功能都可以在Windows的所有版本或版本中提供。系统可能需要升级和/或单独购买硬件,驱动程序,软件或BIOS更新,以充分利用Windows功能。Windows 11自动更新,始终启用。ISP费用可能会适用,并且随着时间的推移可能会适用其他要求。 请参阅http://www.windows.com。 Windows 11 In S模式下的Windows 11专门与Windows中Microsoft Store的应用程序合作。 无法更改某些默认设置,功能和应用程序。 某些与Windows 11兼容的配件和应用程序可能不起作用(包括某些防病毒,PDF作者,驱动程序实用程序和可访问性应用程序),并且性能可能会有所不同,即使您切换出S模式。 如果您切换到Windows 11,则无法切换回S模式。 在Windows.com/smodefaq上了解更多信息。 (2a)多核旨在提高某些软件产品的性能。 并非所有客户或软件应用程序都必须从使用此技术中受益。 性能和时钟频率将根据应用程序工作负载以及您的硬件和软件配置而有所不同。 AMD的编号不是时钟速度的测量。 AMD和Radeon是高级Micro Devices,Inc。的商标(2D)最大Boost时钟频率性能取决于硬件,软件和整体系统配置。 GHz是指处理器的内部时钟速度。 除时钟速度外,其他因素可能会影响系统和应用程序性能。 试用期间需要订阅续订。ISP费用可能会适用,并且随着时间的推移可能会适用其他要求。请参阅http://www.windows.com。Windows 11 In S模式下的Windows 11专门与Windows中Microsoft Store的应用程序合作。无法更改某些默认设置,功能和应用程序。某些与Windows 11兼容的配件和应用程序可能不起作用(包括某些防病毒,PDF作者,驱动程序实用程序和可访问性应用程序),并且性能可能会有所不同,即使您切换出S模式。如果您切换到Windows 11,则无法切换回S模式。在Windows.com/smodefaq上了解更多信息。(2a)多核旨在提高某些软件产品的性能。并非所有客户或软件应用程序都必须从使用此技术中受益。性能和时钟频率将根据应用程序工作负载以及您的硬件和软件配置而有所不同。AMD的编号不是时钟速度的测量。AMD和Radeon是高级Micro Devices,Inc。的商标(2D)最大Boost时钟频率性能取决于硬件,软件和整体系统配置。GHz是指处理器的内部时钟速度。除时钟速度外,其他因素可能会影响系统和应用程序性能。试用期间需要订阅续订。(8A)MCAFEE已预装在您的PC上,并且需要一个帐户才能激活。VPN功能在印度,中国,叙利亚或朝鲜不可用。mcafee在S模式下与Windows 11不兼容。您需要永久切换出S模式。无需切换S模式,但是您将无法重新打开它。MCAFEE和MCAFEE徽标是美国和其他国家的McAfee,Inc。的商标或注册商标。(9)无线访问点和Internet服务需要并单独出售。公共无线访问点有限公司的可用性。(14)共享视频内存(UMA)使用总系统内存的一部分进行视频性能。专用于视频性能的系统存储器无法用于其他程序的其他使用。(15)实际格式的容量较小。内部存储的部分保留用于预加载内容。(19E)无线访问点和Internet服务需要并单独出售。公共无线访问点有限公司的可用性。Wi-Fi 6与先前的802.11规格兼容。仅在支持Wi-Fi 6的国家 /地区可用。(20)所有绩效规格代表惠普组件制造商提供的典型规格;实际性能可能会更高或更低。(21)实际的电池瓦小时(WH)将因设计能力而异。电池容量自然会随着保质期,时间,使用,环境,温度,系统配置,加载应用程序,功能,电源管理设置和其他因素而自然降低。(22)从注册之日起一年的100GB免费在线存储。有关完整的详细信息和使用条款,包括取消政策,请访问dropbox.com的网站。需要互联网服务,不包括。(23)HP BIOS保护与Windows 11一起使用,该Windows与您的PC发货。(25)Bluetooth®5.3操作需要Microsoft OS/Chrome OS支持。直到获得Microsoft OS/Chrome OS支持,Bluetooth®5.3将以Bluetooth®5.2或更低的速度发挥作用。(26)Bluetooth®是其所有人拥有的商标,并由HP Inc.使用许可。(27)基于IEEE 1680.1-2018Epeat®的U.Sepeat®注册。epeat®地位随国家而异。有关更多信息,请访问www.epeat.net。(39)查看FHD图像需要完整的高清(FHD)内容。(40)活跃的不活动观看区域到主动观看区域和边框的非活动观看区域。用垂直盖在桌子上的盖子。(41)需要Windows11。某些功能需要NPU。功能交付和可用性的时机因市场和设备而异。需要Microsoft帐户才能登录。如果不可用的副本,副标题将导致Bing搜索引擎。请参阅http://aka.ms/windowsaifeatures。(42)USB Type-C®和USB-C®是USB实施者论坛的商标。(53)致电1.800.474.6836或支持。在标准保修期满后,可能会收取事件费。排除塑料袋和塑料泡沫板。(56)有关更多信息,请访问hp.com/go/hpsupportassistant。(链接在美国以外的链接将有所不同)HP支持助手仅在基于Windows的PC上可用。更新和连接到HP支持所需的Internet连接。(58b)基于HP内部测试,使用CrystalDiskmark基准测试软件。性能依次更快(仅读取)。(61)无闪烁的功能正在通过整合DC降低LED背光或LED排放来消除屏幕闪烁。OLED面板在100nits以上时具有DC降低功能。(62)能源之星和能量之星标记是美国环境保护局拥有的注册商标。(63b)钥匙扣中包含的消费后再生塑料的百分比因产品而异。(63C)每个组件中包含的海洋结合塑料百分比因产品而异。(63E)100%外盒/瓦楞纸垫包装,由可持续采购的认证和再生纤维制成。由100%再生木纤维和有机材料制成的纤维垫。(75)由HP使用连续FHD视频播放测试的电池寿命,1080p(1920x1080)分辨率,200 Nits亮度,系统音频水平为图像默认值,播放器音频水平为100%,播放了来自本地存储,固定或通过扬声器的本地存储,扬声器(如果没有辅助式插孔)的全屏幕(如果无需连接)。实际电池寿命会因配置而定,最大容量自然会随时间和使用而降低。(76)重量和系统尺寸可能由于配置和制造方差而波动。(78)在系统关闭时45分钟内将电池充满高达50%(使用“关闭”命令)。建议与笔记本上提供的HP适配器一起使用,不建议使用较小的容量电池充电器。充电达到50%的容量后,充电速度将恢复正常速度。充电时间可能因系统公差而变化+/- 10%。(80)HP使用连续的Netflix视频播放测试的电池寿命,Microsoft Edge浏览器,200 Nits亮度,系统音频级别作为图像默认值,连接的耳机或通过扬声器(如果没有音频插孔端口)播放全屏幕,无线,无线。实际电池寿命会因配置而定,最大容量自然会随时间和使用而降低。(89)功能可能需要软件或其他第三方应用程序来提供所述功能。需要互联网服务,不包括。(89b)摄像机分辨率是指图像传感器活动像素。实际映像捕获像素和纵横比取决于所选的应用。实际产品可能与数据表上显示的图像有所不同。©版权所有2024 HP Development Company,L.P。本文包含的信息如有更改,恕不另行通知。在此类产品和服务随附的明确保修陈述中阐明了HP产品和服务的唯一保证。没有任何解释为构成额外的保修。HP不对此处包含的技术或编辑错误或遗漏不承担任何责任。所有其他商标都是其各自所有者的财产。Microsoft和Windows是美国和/或其他国家的Microsoft Corporation的注册商标或商标。08/22/2024_r3 kc

期刊 - HP 实验室
模拟示波器在实验室分析应用中几乎被数字或数字化示波器所取代,但它却拒绝消亡。由于其成本低、控制简单易用和实时显示,它仍然是工程师和技术人员进行故障排除的首选。将此视为一项挑战,惠普科罗拉多斯普林斯分部的工程师着手设计一款数字化示波器,故障排除人员不仅会发现它与模拟示波器相当,而且实际上会更喜欢它。HP 54600 系列数字化示波器具有通常与最常用于故障排除的全功能 100 MHz 模拟示波器相关的所有功能。它们具有相同的带宽 - 它们是 MHz - 并且在成本和易用性方面具有可比性。虽然它们显然是连续示波器(显示的波形由点而不是连续的线组成),但 HP 调整系列示波器在大多数情况下对电路调整的响应速度与模拟示波器一样快,实际上在某些任务上表现更好。使它们优于模拟示波器(数字化示波器可与之媲美)的原因是只有数字化示波器才能提供的存储和测量功能阵列。由于波形数据是在内存中采样和存储的,因此可以在触发事件之前和之后查看数据,以数学方式处理数据,并以衰减的方式无限期地显示波形。从第 6 页的介绍性文章开始,到与模拟示波器进行故障排除的正面比较(第 57 页)结束,本期共有 9 篇文章涉及 HP 54600 系列示波器的设计。它们描述了如何通过高水平的电路集成、使用表面贴装技术装载印刷电路板、经济高效的机械封装以及对制造过程的精心关注(包括测试专用和测试设备的成本)来解决成本问题。通过为主要控制功能提供专用旋钮而不是菜单驱动的软键用户界面来解决易用性问题,尽管保留了菜单和软键来控制数字化示波器功能。通过采用新架构和两个专用集成电路,显示速率能力提高到每秒一百万点,是其他数字化示波器的五十到一百倍。通过将每条轨迹显示的点数增加四倍,波形平滑度得到改善。您可以在文章的第 11 页找到有关架构和定制 IC 的详细信息,在第 36 页找到有关机械设计的详细信息,在第 21 页找到有关测试策略和测试系统的详细信息。验证而非特性分析的测试策略大大减少了需要测量的参数数量,而新的基于 FFT 的测量算法(第 29 页)进一步改进了仅使用数字万用表的生产测试系统。在第 41 页,您可以阅读有关确保 HP 54600 系列示波器符合电磁兼容性国际和军用标准(对于故障排除仪器而言非常重要)的步骤。第 45 页的文章介绍了一种使用数字化示波器的存储和无限持久性能力的新方法。它被称为自动存储,以全强度显示最新效果,以半强度显示较早的轨迹,以便用户可以更轻松地看到调整的效果。HP 54600 系列和其他 HP 数字化示波器中使用的模数转换器是 16 通道、16 位、间接类型(第 48 页)。除了将波形样本转换为数字数据外,它还用于校准垂直增益。
期刊 - HP 实验室
模拟示波器在实验室分析应用中几乎已被数字或数字化示波器所取代,但它却拒绝消亡。由于其成本低、控制简单、显示实时,它仍然是工程师和技术人员进行故障排除的首选。惠普科罗拉多斯普林斯分部的工程师们将此视为一项挑战,着手设计一种数字化示波器,故障排除人员不仅会发现它与模拟示波器相当,而且实际上更喜欢它。HP 54600 系列数字化示波器具有通常与最常用于故障排除的全功能 100 MHz 模拟示波器相关的所有功能。它们具有相同的带宽 - 它们是 MHz - 并且在成本和易用性方面相当。虽然它们显然是连续示波器(显示的波形由点而不是连续的线组成),但 HP 调整系列示波器在大多数情况下对电路调整的响应速度与模拟示波器一样快,而且实际上在某些任务上表现更好。与模拟示波器相比,数字化示波器更受欢迎的原因在于只有数字化示波器才能提供的存储和测量功能。由于波形数据是在内存中采样和存储的,因此可以在触发事件之前和之后查看数据,以数学方式处理数据,并无限期地显示带有衰减的波形。从第 6 页的介绍性文章开始,到与模拟示波器的正面比较(用于故障排除)(第 57 页),本期共 9 篇文章讨论了 HP 54600 系列示波器的设计。他们描述了如何通过高水平的电路集成、使用表面贴装技术装载印刷电路板、具有成本效益的机械封装以及对制造过程的精心关注(包括专用测试和测试设备的成本)来解决成本问题。通过为主要控制功能提供专用旋钮而不是菜单驱动的软键用户界面,部分解决了易用性问题,尽管保留了菜单和软键来控制数字化示波器功能。通过新的架构和两个专用集成电路,显示速率能力提高到每秒一百万点,是其他数字化示波器的五十到一百倍。通过将每条轨迹显示的点数增加四倍,波形平滑度得到了改善。您将在第 11 页的文章、第 36 页的机械设计以及第 21 页的测试策略和测试系统中找到架构和定制 IC 的详细信息。验证而不是特性的大量测试策略大大减少了需要测量的参数数量,和新的基于 FFT 的测量算法(第 29 页)进一步改进了生产测试系统部分为内置式,只使用两个信号源和一个外部数字万用表。在第 41 页,您可以阅读有关确保 HP 54600 系列示波器符合电磁兼容性国际和军用标准的步骤——这对于故障排除仪器很重要。第 45 页的文章介绍了一种使用数字化示波器的存储和无限持久能力的新方法。它称为自动存储,以全强度显示最新效果,以半强度显示早期轨迹,以便用户更容易看到调整的效果。HP 54600 系列和其他 HP 数字化示波器中使用的模数转换器是 16 通道、16 位、间接类型(第 48 页)。除了将波形样本转换为数字数据之外,它还用于校准垂直增益。

HP SitePrint机器人快速设置HP安全经理HP安全经理
最常用的日志文件用于故障排除设备特定补救问题EAPDEVICELIB.LOG,EAPNETWORKLIB.LOG和HPSM_SERVIE.LOG。为了解决每晚维护的故障排除问题,需要维护Enancetask.log。对于即时解决问题的故障排除问题,需要instanton.log。用于解决任务的故障排除问题,HPSM_Service.log以及最终eapdevice.lib.log和eapnetworklib.log。用于对设备证书进行故障排除评估/补救问题,需要HPCm.LOG,最终eapdevice.lib.log和eapnetworklib.log。需要使用scep scep.log和hpcm.logs使用设备证书进行故障排除评估/补救问题,最终需要eapdevice.lib.log和eapnetworklib.log。需要使用EST.LOG和HPCM.LOGS进行故障排除评估/补救问题,最终需要eapdevice.lib.log和eapnetworklib.log。

2023 HP建筑形式S.xlsx
至少(1)便携式灭火器应在厨房10英尺范围内。灭火器应列出,不小于2a:10b:c的住宅额定值不超过10磅。灭火器的悬挂在地板上方不超过5个必须可见的,易于访问且附近的出口位置。

六辆卡车 - 4621 HP!
舒适性 - 毫无疑问,至少部分是由于超长的轴距以及R-CAB的示例性绝缘材料,这意味着Scania在其电气版本中具有最高的电动版本,就像其燃烧型号一样,并且在驾驶员等级中得分很高(请参阅第31页)。沃尔沃FH电气的脚后跟很热。好吧,它的设置可能不是最先进的,但是方向盘后面的驾驶员不在乎这一点 - 可能从低范围来看。i-Shift换档平稳,快速地更换了齿轮,标准的空气弹头前轴为几乎像教练一样的骑行舒适度构成了贡献,沃尔沃还为噪音绝缘的最高标记得分。这绝不是说其他四名参赛者也没有提供世界上没有com bution Engine的安静程度


违禁品缴获量 - HP - 12.03
3. 如果发现货币,但无人认领,则成员将扣押该货币,无论金额多少,可将其作为无人认领或遗弃的财产、无人认领的证据处理,或通过没收追回。成员必须确保任何持有货币的人在否认对货币的所有权并放弃对货币的任何认领后,填写资产认领豁免和扣押通知权利豁免表格 (HSMV 61076) 或记录此类拒绝。在可能被没收的情况下发现货币时,必须联系主管和部队法律顾问。
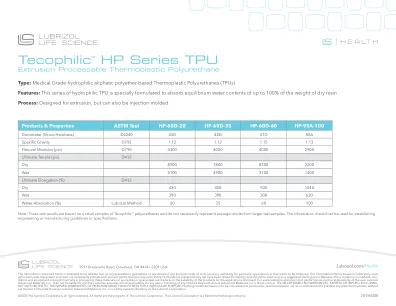
产品规范
本文所包含的信息被认为是可靠的,但没有任何形式的陈述,担保或保证就其准确性,适用于特定申请或要获得的结果。这些信息通常基于实验室的小型设备,不一定表明最终产品性能或可重现性。提出的配方可能没有进行稳定性测试,仅应作为建议的起点。由于在处理这些材料时商业上使用的方法,约束和设备的变化,因此没有对产品适用于披露的申请的适用性。全尺度测试和最终产品性能是用户的责任。Lubrizol Advanced Materials,Inc。不承担任何责任,并且客户对除Lubrizol Advanced Materade,Inc。的直接控制外的任何用途或处理任何材料都承担所有风险和责任。卖方不对明示或暗示的担保,包括但不限于对特定目的的适销性和适合性的隐含保证。本文中没有任何包含在未经专利所有者许可的情况下练习任何专利发明的授权,也不应将其视为诱因。Lubrizol Advanced Materials,Inc。是Lubrizol Corporation的全资子公司。