XiaoMi-AI文件搜索系统
World File Search Systemoligo

magmax™核心核酸纯化试剂盒
描述Thermo Scientific™First Strand cDNA合成试剂盒是一个完整的系统,用于有效合成mRNA或总RNA模板的第一链cDNA。该试剂盒使用M-Mulv逆转录酶,与AMW逆转录酶相比,RNase H活性较低。酶在37°C下保持活性,适合于9 kb的cDNA合成。套件提供的重组Thermo Scientific™Ribolock™RNase抑制剂可有效保护RNA在高达55°C的温度下免于降解。该套件均配有寡核(DT)18和随机己酯引物。随机六聚体引物与非特异性结合,并用于合成总RNA种群中所有RNA的cDNA。寡(DT)18底漆选择性地向poly(a)RNA的3末端退火,仅从poly(a)尾mRNA中合成cDNA。基因特异性底漆也可以与试剂盒一起使用,以从指定序列中进行质合。与该系统合成的第一链cDNA可以直接用作PCR或实时PCR中的模板。它也是第二链cDNA合成或线性RNA扩增的理想选择。可以将放射性和非放射性标记的核苷酸纳入第一个链cDNA,以用作包括微阵列在内的杂交实验的探针。

SYNTAX 96 高保真套装
SYNTAX 96 高保真试剂盒包括并行酶促合成 96 个即用型 DNA 寡核苷酸(长度可达 120 个核苷酸 (nt))所需的试剂和耗材,适用于需要高序列准确性的应用,例如基因组装、蛋白质诱变或 CRISPR 基因编辑。寡核苷酸的快速设置和打印可在下游工作流程中当天或次日使用。使用 SYNTAX 系统进行寡核苷酸合成时,合成和运行试剂盒都是必需的。

FTIR-ATR检测蛋白质和小分子通过DNA结合 前额叶皮层在持久性中的作用 距离距离协议 加强学习中的概括 结合质粒DNA转移在幽门螺杆菌幽门螺杆菌中,由染色体编码的弛豫酶和trag样蛋白介导 人脑功能的发展 冒险参加青春期:大脑和行为科学的新观点 战略规划在现代组织中的作用Marilen Pirtea 1 Cristina Nicolescu 2 Claudiu Botoc 3摘要:有一个非常重要的 Walrasian,Neo-Hobbesian和Marxian模型 心理和同理心理论的神经元相关 生化分析和优化玻璃 - 西硅芯片中抑制和吸附现象 资本主义经济中的失业 隐藏的马尔可夫模型适合文本生成 上生长的gan的gan mishemts and MMIC的性能和可靠性 选定的书目
傅里叶变换红外衰减的总反射(FTIR-ATR)已广泛用于研究表面和界面上的吸附和反应。与其他技术不同,例如荧光,无线电标记和电动检测,FTIR-ATR不需要额外的标签,并且可以提供有关系统的大量信息。因此,FTIR-ATR具有许多潜在的生物学应用,并且有望成为一种高敏感,无标签和通用的生物传感方法。近年来,FTIR-ATR生物学应用的主要研究工作集中在(a)原位观察蛋白质或细胞吸附[1-5]; (b)生物膜的结构和方向分析[5-11]; (c)检查酶促反应[12,13]。我们的兴趣集中于FTIR-ATR的生物传感应用,以检测与固定的DNA或寡核苷酸(Oligo)探针有关的生化过程。

固态NA电池的柔性无溶剂聚合物电解质
基于碱性和碱性地球元素的lIthium后电池是更便宜的技术,其潜力有可能在过渡到更清洁和可持续的能源中的颠覆性变化,从而降低了对化石燃料的依赖。这项贡献涉及钠导电的无溶剂聚合物电解质对钠聚合物电池的发展和表征。通过α,ω-二羟基 - oligo(氧化乙烯)的多浓度与不饱和二甲酰基获得,其进一步的固化会导致无定形的网络电解质膜。在不同的O/Na比下使用NaClo 4和NACF 3 SO 3 SO 3,最佳的聚合物电解质达到90℃的阳离子电导率(σ +),超过1 ms cm -1,而保持机械完整性至少至少120°C. c.
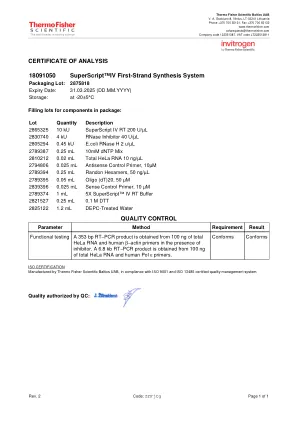
18091050 SuperScript™IV 第一链合成系统
批次 数量 描述 2865325 10 kU SuperScript IV RT 200 U/µL 2830740 4 kU RNase 抑制剂 40 U/µL 2805294 0.45 kU E.coli RNase H 2 u/µL 2789387 0.25 mL 10mM dNTP 混合物 2810212 0.02 mL 总 HeLa RNA 10 ng/µL 2794806 0.025 mL 反义对照引物,10µM 2789394 0.25 mL 随机六聚体,50 ng/µL 2789395 0.05 mL Oligo (dT)20,50 µM 2839396 0.025 mL 正向对照引物, 10 µM 2789374 1 mL 5X SuperScript™ IV RT 缓冲液 2821527 0.25 mL 0.1 M DTT 2825122 1.2 mL DEPC 处理水

利用 ARCUS 巨核酸酶消除乙肝病毒的基因编辑方法
图 3:HBV-ARCUS-POL 核酸酶在各代中表现出更高的特异性 • 含有一个部分整合的 HBV 基因组的 HepG2 细胞被转染了高水平的 HBV-ARCUS- POL 核酸酶以及 DNA“标签”。分离 gDNA 并使用 Oligo Capture NGS 评估脱靶编辑。 • 每个蓝点代表一个潜在的切割位点,X 轴表示恢复的读取次数,点的颜色表示每个位点与预期的 22bp 目标位点相比的错配数。 • 最有可能真实的脱靶位点是那些具有大量读取次数或错配较少的位点。这些包含在黄色轮廓框内。橙色圆圈表示已整合到 HepG2 细胞系基因组中的预期目标位点。

rprimer:一个用于设计...的 R/bioconductor 包
摘要背景:本文介绍了一种新的 R/Bioconductor 包 rprimer,用于设计序列可变病毒的简并寡核苷酸和 PCR 检测。多重 DNA 序列比对用作输入数据,而输出则包括综合表格(数据框)和仪表板式图表。工作流可以直接从 R 控制台或通过图形用户界面(Shiny 应用程序)运行。本文演示并评估了 rprimer,方法是使用它设计两种诺如病毒基因组 I (GI) 检测:一种用于定量检测的 RT-qPCR 检测和一种用于 Sanger 测序和基于聚合酶衣壳的基因分型的 RT-PCR 检测。结果:使用检测为诺如病毒 GI 阳性的粪便样本评估生成的检测。RT-qPCR 检测准确扩增和量化了所有样本,并显示出与广泛使用的标准化检测相当的性能,而 RT-PCR 检测成功测序和基因分型了所有样本。通过与三个类似的免费软件包进行比较,确定了该软件包的优点和局限性。不同工具的几个特点是相似的,但 rprimer 的重要优势在于其速度、寡核苷酸设计的灵活性和可视化能力。结论:开发了一个 R/Bioconductor 软件包 rprimer,并证明它可以成功设计引物和探针,用于定量检测和基因分型序列可变的病毒。该软件包提供了一种高效、灵活且直观的退化寡核苷酸设计方法,因此可以帮助病毒研究和方法开发。

创建和验证人类Implintome的第一个Infinium DNA甲基化阵列
图1。使用荧光团 - 猝灭剂系统对DNA二级结构进行高通量热力学测量。a。折叠(淬火)和展开(荧光)状态的DNA分子的示意图。b。固定在测序芯片表面上的荧光DNA簇的图像。顶部:仅具有荧光团偶联的寡核(CY3),以及荧光团和淬火剂偶联的寡核能的图像。底部:每个图像中DNA分子的示意图。所有图像均标准化为超稳定的茎和重复对照变体,以依赖温度对荧光和淬火的影响,如图S1D。 c。库型和淬灭剂偶联的寡核苷酸的恒定序列结合位点之间的库变体设计。 红色代表每种类型内的支架核苷酸恒定,蓝色可系统排列的变量('n')。 每个类下的数字指示每个类中唯一序列的数量。 d。对照构建体的荧光测量,其中荧光团和淬灭器之间的单链距离在单核苷酸步骤下增加。 橙色线显示理论拟合。 e。在较高的温度(熔体曲线,X轴)和降低温度(退火曲线,Y轴)f的情况下,∆G 37的相关性来自图书馆变体。熔融曲线的代表性示例在GC含量方面有所不同。 g。三个熔体和一个退火曲线实验重复的∆G 37的Pearson相关性。S1D。c。库型和淬灭剂偶联的寡核苷酸的恒定序列结合位点之间的库变体设计。红色代表每种类型内的支架核苷酸恒定,蓝色可系统排列的变量('n')。每个类下的数字指示每个类中唯一序列的数量。d。对照构建体的荧光测量,其中荧光团和淬灭器之间的单链距离在单核苷酸步骤下增加。橙色线显示理论拟合。e。在较高的温度(熔体曲线,X轴)和降低温度(退火曲线,Y轴)f的情况下,∆G 37的相关性来自图书馆变体。熔融曲线的代表性示例在GC含量方面有所不同。g。三个熔体和一个退火曲线实验重复的∆G 37的Pearson相关性。h。各种构造类别的标准误差为∆G 37的函数。

CAS9生成条件小鼠等位基因的方法
1。我们对Yang等人发表的MECP2基因座的结果。已通过Jaenisch(8 - 10%正确的等位基因),Yang(8%正确的等位基因)和Hatada的组(2 - 6%正确等位基因)[3]的独立实验复制。此外,多个同行评审的出版物[3-7]成功使用了此方法来创建条件敲除(CKO)小鼠(在11个基因座中有9个成功,效率为2.5%至18%)。我们注意到,CRISPR/ CAS9生成CKO小鼠的效率可能会有所不同,这可能是由于平台特征或实验条件的不同。2。Gurumurthy等人使用的条件。[1]与我们论文中使用的条件不符。Gurumurthy等人使用的CRISPR试剂的浓度。 '在MECP2基因座上的研究[1](Cas9 mRNA的10 ng/μL,SGRNA的10 ng/μL,寡核素的10 ng/μL)比Yang等人所用的 sgrNA的RNA和10 ng/μL)。 ' s实验(CAS9 100 ng/μL,SGRNA 50 ng/μL和100 ng/μL的实验)[2]和Yang等。 ' s先前[8]和以下出版物[9-12]。 众所周知,CRISPR试剂的浓度与基因组编辑效率密切相关。 3。 我们在原始论文中使用了压电驱动的合子注入方法,该方法允许以更高的浓度注入CRISPR组件。 Gurumurthy等人使用的该方法和前核注射方法之间的差异。 也可能有助于成功的利率差异。sgrNA的RNA和10 ng/μL)。 's实验(CAS9 100 ng/μL,SGRNA 50 ng/μL和100 ng/μL的实验)[2]和Yang等。 's先前[8]和以下出版物[9-12]。众所周知,CRISPR试剂的浓度与基因组编辑效率密切相关。3。我们在原始论文中使用了压电驱动的合子注入方法,该方法允许以更高的浓度注入CRISPR组件。Gurumurthy等人使用的该方法和前核注射方法之间的差异。也可能有助于成功的利率差异。

生物信息学的基础
1。什么是生物信息学,基因组测序项目,模型生物,序列 - 结构 - 功能,生物信息学研究所,生物信息学和转录组,蛋白质组,代谢组,基本序列信息。生物数据库,数据格式,查询形式。比较2个序列,氨基酸相似性,相似性表,相似性因子,数据库中的相似性搜索,FASTA和BLAST算法,期望值。阅读和处理序列数据(Chromas)的方法。准备限制地图(从浮雕包中重新包装程序)。使用来自“浮雕”软件包(绘图ORF,显示ORF和GET ORF)的应用程序读取帧。基于核苷酸序列(来自浮雕封装的Transeq程序)基本序列数据库(DDBJ,EMBL,GenBank)生成蛋白质序列。蛋白质序列数据库。基因组浏览器。通过Expasy Server,数据库:瑞士蛋白石,Prosite访问各种生物信息来源。底漆设计,基本和高级参数,程序:Oligo,ePrimer3(浮雕)),Prime(GCG)。