XiaoMi-AI文件搜索系统
World File Search Systemorbitals

核壳模型中原子核结构的量子纠缠模式
摘要 量子纠缠为研究原子核等强相关系统的底层结构提供了独特的视角。在本文中,我们使用量子信息工具分析核壳模型中轻和中等质量的铍、氧、氖和钙同位素的结构。我们对壳模型价空间的不同均分采用不同的纠缠度量,包括单轨道纠缠、互信息和冯诺依曼熵,并确定与核单粒子轨道的能量、角动量和同位旋相关的模式纠缠模式。我们观察到单轨道纠缠与价核子的数量和壳层的能量结构直接相关,而互信息则突显了质子-质子和中子-中子配对的迹象。质子和中子轨道在所有测量中都是弱纠缠的,事实上,在所有可能的价态空间均分中,它们的冯·诺依曼熵最低。相反,具有相反角动量投影的轨道具有相对较大的熵。这一分析为设计更高效的量子算法以应对嘈杂的中尺度量子时代提供了指导。

消除 APW/LAPW 基组中的线性化误差
详细介绍了使用 APW/LAPW 类型基组以及由局部轨道提供的灵活扩展来实现相对论密度泛函理论 (RDFT) 方程的方法。使用完全相对论方法和 α -U 作为示例,证实了先前发现的 APW/LAPW 基组的高导数局部轨道 (HDLO) 扩展对于提高 DFT 计算精度的重要性。高能局部轨道 (HELO) 对 GW 计算来说必不可少,但在提高 DFT 应用精度方面却效率低得多。结果表明,对于本文考虑的五种材料的电子自由能,采用一种简化的相对论效应方法,即仅考虑它们在 muffin-tin (MT) 球体内部,会产生基本相同的结果(与完全相对论方法相比)。通过比较简化方法对电子自由能的影响和对电子动能的影响,我们得出结论,自由能对我们描述间隙区域的相对论效应的方式的不敏感性与该量的变分性质有关。

在压力下LA3NI2O7中的层间耦合驱动的高温超导性
在压力下,新发现的LA 3 Ni 2 O 7中新发现的高温超导性吸引了很多关注。表征电子特性的基本要素是双层NiO 2平面,该平面是通过中间氧原子的3 d Z 2轨道的层间键合结合的。在强耦合极限中,低能物理学由内征抗磁性自旋交换相互作用j K在3 d x 2-y 2轨道之间的j k和3 d z 2轨道之间的层间j k之间描述。考虑到每个站点上的规则并整合了3 d Z 2自由度的自由度,该系统将基于3 d x 2 -2 -y 2轨道的单轨道双层t -j模型还原为单轨道双层T -J模型。通过采用奴隶玻色子方法,求解了键合和配对阶参数的自动一致方程。在物理相关的1 4填充方案附近(掺杂δ¼0。3〜0。5),层间耦合j⊥将常规的单层D-波超导状态调整为S波。强的J⊥可以增强层间超导顺序,从而导致t c急剧增加。有趣的是,可能存在一个有限的制度,在这种制度中,出现了sÞID状态。

发现锂离子电池的铅喹酮阴极材料
fe(ii)自旋跨界(SCO)复合物是分子,其中Fe原子周围的八面体配体场的强度在该领域中,即使温度1-5或磁场的变化,也可以在这些分子中触发旋转状态过渡。9–15在低温下,当T 2G和E G轨道之间的八面体配体场分裂(D OCT)很高时,SCO综合体占据了Diamagnetic(S = 0)低旋转状态(LS)。但是,在温度高于临界过渡温度t c的温度下,当t 2g和e g轨道之间的D OCT降低时,这些分子占据了顺磁性(s = 2)高旋转状态(HS)。14,16–20由于在Fe(II)的SCO复合物以及这些旋转状态的双态性中可以实现此类自旋状态过渡的方便,因此9,21,22这些分子可以使室温旋转的旋转特性构成很好的候选(因为触发了旋转状态过渡的室温,因此很大程度上是可触发旋转状态的过渡,而不是很大程度上是可实现的),并且不可能实现23-25,并且VER且VERIOL无效),并且V-25和23-25。26–28室温磁的存在

修订了客观类型主体能力测试的教学大纲(SAT),以招募化学的讲师(新学校),III级III
修订了客观类型学科能力测试的教学大纲(SAT),以招募招聘,以在高等教育系的化学讲师(学校新)中任职。本文的持续时间为100分。客观类型的主体能力测试(SAT)应涵盖以下主题: - A部分(公共课程和生物化学课程)(60分)无机化学群体理论:群体,对称元素和对称性操作的概念,对点组的分配,对某些无机分子的分配,对乘法的一般繁殖,繁殖,繁殖,繁殖,繁殖,繁殖,繁殖,繁殖,繁殖,繁殖, (矩阵,C 2 V和C 3 V点组的矩阵表示),C 2 V和C 3 V点组的字符和性格表。群体理论在化学键合中的应用(在不同几何和π键的杂交轨道和杂种轨道中的杂交轨道。BF 3,C 2 H 4和B 2 H 6中分子轨道的对称性。 非水溶剂:证明需要非水溶液化学和水作为溶剂的因素是合理的。 硫酸的溶液化学:物理性能,H 2 SO 4中的离子自脱水,高粘度高,高粘度,H 2 SO 4作为酸的化学性,作为脱水剂,作为氧化剂,作为氧化剂,作为一种培养基酸碱中和中性化反应和分化分化的分化的介质。 液体BRF3:物理特性,BRF3中的溶解度,自发,酸碱中和反应,溶解反应和过渡金属氟化物的形成。对称性。非水溶剂:证明需要非水溶液化学和水作为溶剂的因素是合理的。硫酸的溶液化学:物理性能,H 2 SO 4中的离子自脱水,高粘度高,高粘度,H 2 SO 4作为酸的化学性,作为脱水剂,作为氧化剂,作为氧化剂,作为一种培养基酸碱中和中性化反应和分化分化的分化的介质。液体BRF3:物理特性,BRF3中的溶解度,自发,酸碱中和反应,溶解反应和过渡金属氟化物的形成。无机氢化物:分类,制备,粘结及其应用。过渡金属化合物具有键与氢,羰基氢化物和氢化阴离子的键。分类,命名法,韦德的规则,制备,结构和结合在硼氢化物(硼酸盐)和卡顿人中,无机化学中的有机试剂:螯合,螯合,确定螯合物稳定性的因素(环尺寸的效果,金属的氧化状态,金属的氧化状态,金属的氧化状态);在分析中使用以下试剂的使用:二甲基乙二醇(在分析化学中)EDTA(在分析化学和化学疗法中)8-羟基喹啉(在分析化学和化学疗法中)1,10-苯磺烷oltholine(分析化学和化学疗法)(在分析化学和化学疗法中)硫代化学疗法(分析性化学疗法)(分析性化学疗法)(分析性化学方法)(分析)INAICONES(分析)Dithiaz iniazon(分析)Dithiace(分析)Dithiace(分析)Dithiace(Inalistical Chemantication)(分析性化学疗法)Dithiazon(Dithiace)Dithiazone(分析性化学疗法)。金属配体键合-I:晶体场理论的概括,包括在不同环境中脱落D-轨道,影响晶体场分裂大小的因素,结构效应(离子半径,Jahn-Teller效应),热力学效应,晶体场理论的热力学效应(结合,水合和晶格理论),晶体理论,晶体理论,晶体理论,晶体范围,ACFTINE-CRYSTAL TROPDAL-IDECTINE-CRYSTAL IDECTAL IDECTAL IDECTAL IDECTAL-IDECTIND CRYSTAL TROPDAL-FRYSID-ACFTINE-ACFTINE-ACFTINE-FRYSILID(ACFIDINE)在复合物中,用于八面体,四面体和方形平面复合物(不包括数学处理)的分子轨道理论原子光谱:原子中的能级,轨道角动量的耦合,旋转角臂的耦合,旋转角矩,旋转Orbit Orbit,Spin Orbit coupling,Spib Orbit P2案例,

硅纳米带中一维狄拉克费米子的观测支持信息
为了阐明 SiNRs/Ag(110) 中 1D 狄拉克带的起源,我们将 SiNRs/Ag(110) 的展开能带结构投影到不同的原子层,如图 S4(a)-S4(d) 所示。可以看出,狄拉克带主要位于表面 Si 层,在最顶层的 Ag 层只有少量的剩余信号。最顶层 Ag 层中的剩余信号表示 Si 和 Ag 之间的有限能带杂化。第 8 个 Ag 层仅包含 Ag(110) 的体能带,如图 S4(c) 所示。通过比较图 S4(a) 和 S4(c),我们还可以得出结论,狄拉克带附近强度较高的能带来自 Ag(110) 的体能带。事实上,由于我们计算中的平板几何形状,这些能带来自 Ag 体 sp2 能带的子能带。为了研究狄拉克能带的轨道组成,我们将展开的能带结构投影到 Si s 和 Si ad 原子的不同轨道上,如图 S4(e)-S4(l) 所示,发现狄拉克能带主要由 Si spz 轨道组成。这些结果与我们的 TB 分析结果一致,即 Si s 和 Si ad 原子的 pz 轨道是解耦的。
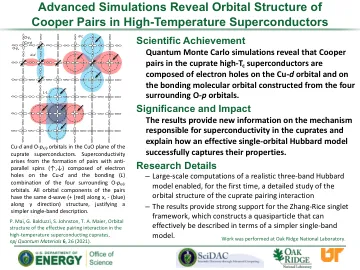
高级模拟揭示高温超导体中库珀对的轨道结构
铜氧化物超导体 CuO 平面中的 Cu- d 和 O- px/y 轨道。超导性源于 Cu- d 上的电子空穴与周围四个 O- px/y 轨道的键合 ( L ) 组合形成的具有反平行自旋 (↑,↓) 的对。这些对的所有轨道分量都具有相同的 d 波 (+ (红色) 沿 x 方向,- (蓝色) 沿 y 方向) 结构,从而证明了更简单的单波段描述。
![arXiv:2002.07901v1 [quant-ph] 2020 年 2 月 18 日](/simg/c\c194b283812d90cda7068798de2ac8669e964e6a.webp)
arXiv:2002.07901v1 [quant-ph] 2020 年 2 月 18 日
量子计算是一种新的计算范式,有望有效模拟量子力学系统。然而,与工业相关的分子尺寸相比,嘈杂的中型量子 (NISQ) 设备提供的硬件范围仍然很小。本文引入了增量法 (MI),以帮助加快 NISQ 设备在量子化学模拟中的应用。MI 方法将分子系统的电子关联能量表示为轨道、原子、分子或碎片的截断多体展开。在这里,系统的电子关联以占据轨道的形式展开,并采用 MI 方法系统地减少占据轨道空间。同时,虚拟轨道空间基于冻结自然轨道 (FNO) 减少,FNO 是使用二阶多体微扰理论的单粒子密度矩阵获得的。这样,构建了一种称为 MI-FNO 方法的方法,用于系统地减少量子化学模拟中的占用空间和虚拟空间。然后可以通过任何算法(包括相位估计算法和变分量子特征值求解器等量子算法)求解由 MI-FNO 减少引起的子问题,以预测分子系统的相关能量。在 cc-pVDZ 基组内,针对小分子(即 BeH 2 、CH 4 、NH 3 、H 2 O 和 HF)的情况,研究了 MI-FNO 方法的准确性和可行性。然后,使用对工业相关的中型催化剂分子(“受限几何”烯烃聚合催化剂)的量子比特计数估计,研究了所提出的框架对于实际工业应用中使用的较大分子的有效性。我们表明,即使采用适度截断虚拟空间,MI-FNO 方法也能将量子比特需求减少近一半。这样一来,我们的方法可以促进基于较小但更现实的化学问题的硬件实验,从而有助于表征 NISQ 设备。此外,降低量子比特需求有助于扩大可在量子化学应用中模拟的分子系统的大小,从而大大增强大规模工业应用的计算化学研究。

具有大自旋间隙的二维半金属的设计
二维半金属在磁性纳米器件中展现出巨大的潜力。然而,二维半金属的发现仍然基于逐案评估。本文,我们提出了设计具有大自旋间隙的二维过渡金属基半金属的一般规则,即找到具有洪特规则分裂的 d 轨道和深阴离子 p 轨道能级以使 dp 相互作用最小化的材料。基于对具有扭曲四面体晶场的 54 种过渡金属化合物 MX 2(M = 3 d 区过渡金属;X = VIA-VIIA 元素)的 DFT 计算,我们发现所有铁磁化合物都表现出半金属性。我们将半金属性归因于具有弱 dp 轨道杂化的 M 阳离子的部分填充 d 轨道的洪特规则分裂。由于 Cl p 轨道能级较深(− 8.4 eV),氯化物表现出大于 4 eV 的自旋间隙。我们在过渡金属三氯化物 M Cl 3(M = 3 d 区过渡金属)中验证了这一规则。利用这一规则,我们预测铁磁单层 M Cl 和 M 3 Cl 8(M = 3 d 区过渡金属)是具有大带隙的半金属。这项工作丰富了二维半金属的种类,并可能带来新型磁性纳米器件。

石墨烯和2D材料的简介作业问题集1 29.04.2024
kagome Lattices具有有趣的晶体结构,并具有三分为基础,重复以填充晶体晶格。在图2中,组成单位细胞的三个原子以红色(R),绿色(G)和蓝色(B)标记,以及将最近邻居连接为⃗δ1,⃗δ2和⃗δ3的载体。我们将计算此Kagome晶格的电子带结构。为简单起见,让我们假设原子R,G和B是相同类型的元素,并且电子在最近邻居的P Z轨道之间跳跃。