XiaoMi-AI文件搜索系统
World File Search Systemp38

CHF6297:一种具有鲁棒抗炎活性的新型有效和选择性p38 MAPK抑制剂,适合吸入肺部给药
抑制p38促丝分裂原激活蛋白激酶(MAPKS)是治疗急性和慢性肺炎性疾病的潜在治疗方法。 在这里,我们报告了CHF6297抗炎性作用的体外和体内表征,这是一种新型有效和选择性的p38α抑制剂,旨在吸入递送作为干粉配方。 CHF6297已被证明可以抑制p38α的酶活性,其亚纳摩尔效力(IC 50 = 0.14±0.06 nm),对p38γ和p38Δ的选择性> 1,000倍。 用脂多糖(LPS)以及用TNF-α或香烟烟雾提取物(CSE)刺激的人支气管上皮细胞(BEAS2B)刺激的人外周血单核细胞(PBMC)(PBMC),CHF6297,CHF6297抑制了blow insleukin(IL)-8 low low nOmolor potence。 CHF6297通过使用仅鼻子吸入装置作为微塑料干粉配方,与乳糖剂量依赖性地抑制了LPS诱导的中性粒细胞在支气管肺泡灌洗液(BALF)中抑制LPS诱导的中性粒细胞 - 抑制LPS诱导的嗜中性粒细胞 - 抑制LPS诱导的嗜中性粒细胞 - 抑制LPS诱导的中性粒细胞 - CHF6297。 chf6297对大鼠的剂量施用剂量依赖性地对抗IL-1β(0.3 mg/kg)诱导的嗜中性粒细胞中的嗜中性粒细胞(ED = 0.22 mg/kg),在BALF中IL-6水平增加(ED 50 = 0.82 mg/kg)。 在暴露于烟草烟雾(TS)的小鼠中,chf6297,鼻内给药(I.N.) 在0.03或0.3 mg/kg时持续4天,剂量依赖性地抑制了BALF中的抗皮质类固醇TS诱导的嗜中性粒细胞中的嗜中性粒细胞。 在炎热的鼠类粉尘螨(HDM)模型中,哮喘病毒A(IAV)(H3N3)(H3N3),CHF6297(0.1 mg/kg,i.n.)抑制p38促丝分裂原激活蛋白激酶(MAPKS)是治疗急性和慢性肺炎性疾病的潜在治疗方法。在这里,我们报告了CHF6297抗炎性作用的体外和体内表征,这是一种新型有效和选择性的p38α抑制剂,旨在吸入递送作为干粉配方。CHF6297已被证明可以抑制p38α的酶活性,其亚纳摩尔效力(IC 50 = 0.14±0.06 nm),对p38γ和p38Δ的选择性> 1,000倍。用脂多糖(LPS)以及用TNF-α或香烟烟雾提取物(CSE)刺激的人支气管上皮细胞(BEAS2B)刺激的人外周血单核细胞(PBMC)(PBMC),CHF6297,CHF6297抑制了blow insleukin(IL)-8 low low nOmolor potence。CHF6297通过使用仅鼻子吸入装置作为微塑料干粉配方,与乳糖剂量依赖性地抑制了LPS诱导的中性粒细胞在支气管肺泡灌洗液(BALF)中抑制LPS诱导的中性粒细胞 - 抑制LPS诱导的嗜中性粒细胞 - 抑制LPS诱导的嗜中性粒细胞 - 抑制LPS诱导的中性粒细胞 - CHF6297。 chf6297对大鼠的剂量施用剂量依赖性地对抗IL-1β(0.3 mg/kg)诱导的嗜中性粒细胞中的嗜中性粒细胞(ED = 0.22 mg/kg),在BALF中IL-6水平增加(ED 50 = 0.82 mg/kg)。 在暴露于烟草烟雾(TS)的小鼠中,chf6297,鼻内给药(I.N.)CHF6297。chf6297对大鼠的剂量施用剂量依赖性地对抗IL-1β(0.3 mg/kg)诱导的嗜中性粒细胞中的嗜中性粒细胞(ED = 0.22 mg/kg),在BALF中IL-6水平增加(ED 50 = 0.82 mg/kg)。在暴露于烟草烟雾(TS)的小鼠中,chf6297,鼻内给药(I.N.)在0.03或0.3 mg/kg时持续4天,剂量依赖性地抑制了BALF中的抗皮质类固醇TS诱导的嗜中性粒细胞中的嗜中性粒细胞。在炎热的鼠类粉尘螨(HDM)模型中,哮喘病毒A(IAV)(H3N3)(H3N3),CHF6297(0.1 mg/kg,i.n.)与已处理的IAV/HDM挑战小鼠相比,气道中性粒细胞显着降低。将CHF6297以无效的本质(0.03 mg/kg)的效果加入布德索尼德时,它增强了类固醇的抗炎性作用。Overall, CHF6297 effectively counteracted lung in fl ammation in experimental models where corticosteroids exhibit limited anti-in fl ammatory activity, suggesting a potential for the treatment of acute exacerbations associated with chronic obstructive pulmonary disease (COPD) and asthma, acute lung injury (ALI), and viral-induced hyperin fl ammation.

出版商更正:疟原虫色素通过激活人类 iPSC 衍生的神经元中的 DNA 损伤、p38 MAPK 和神经退行性通路诱发疟疾
开放存取本文采用知识共享署名4.0国际许可证,允许以任何媒体或格式使用、共享、改编、分发和复制,只要您给予原作者和来源适当的信任,提供知识共享许可证的链接,并表明是否做了更改。本文中的图像或其他第三方资料包含在文章的知识共享许可证中,除非在资料的致谢中另有说明。如果资料未包含在文章的知识共享许可证中,并且您的预期用途不被法定规定允许或超出了允许的用途,您将需要直接从版权所有者处获得许可。要查看此许可证的副本,请访问http://creativecommons.org/licenses/by/4.0/。

开发小分子 Losmapimod 来调节基因表达,治疗 FSHD 的根本原因
• 使用专有探针库确定目标 • Fulcrum 产品引擎将 p38α (MAPK) 确定为 DUX4 表达的关键调节器 • 使用基因组学和化学基因组学工具在多个细胞中验证 p38α (MAPK) • 确定可降低 DUX4 表达的化合物 • 使用患者来源的肌管建模疾病 • 第 1 阶段:临床安全性、耐受性、剂量 (PK) • 第 2 阶段:临床概念验证 • 第 3 阶段:临床益处确认
大学歌手
有丝分裂原激活的蛋白激酶激酶激酶4(MAP3K4)通过调节胰岛素样生长因子1受体1(IGF1R),胰岛素受体(IR)(IR)和AKT信号通路来促进胎儿和胎盘生长[1]。MAP3K4激酶失活(KI)降低IGF1R/IR表达和活性以及AKT激活,从而减少胎儿和胎盘生长[1]。map3k4 ki也会导致胎儿骨骼缺陷[2]。迷宫胎盘层可实现母体和胎儿血之间的气体,营养和废物交换[3]。胎盘是通过滋养细胞(TS)细胞的分化而形成的,并通过去除促进干细胞状态的因子导致在迷宫中发现的滋养细胞形成的因子来形成这些细胞[1]。MAP3K4激酶激活还促进了其他蛋白质的激活,包括p38促丝分裂原激活的蛋白激酶(MAPK)信号通路,在TS细胞中[4]。然而,MAP3K4激酶活性在分化滋养细胞中p38信号通路调节中的作用尚不清楚。该项目的目标是(1)定义MAP3K4激酶激活在迷宫滋养细胞中p38途径控制p38途径中的作用,并且(2)确定TS细胞分化对迷宫滋养细胞对该途径激活的影响。

原始研究ARHGAP6通过Rhoa-Rock1-p38 MAPK信号促进铁毒性抑制乳腺癌肿瘤的生长
背景:铁铁作用是一种不同的铁细胞死亡形式,是由于活性氧(ROS)的产生引起的严重脂质过氧化引起的。乳腺癌患者的生存与Rho鸟苷三磷酸酶水解酶(GTPase)活化蛋白6(ARHGAP6)的肿瘤抑制特性相关。这项研究研究了ARHGAP6对乳腺癌螺栓吞噬作用的影响和机制。方法:使用定量RT-PCR,Western印迹和免疫荧光染色,在基因表达数据集,癌组织样品和细胞中检测到ARHGAP6表达。ARHGAP6。使用5-乙基-2-脱氧尿苷(EDU)测定法测量细胞增殖,并使用LDH细胞毒性测定法测定细胞死亡率。As indicators of ferroptosis, Fe 2+ ion content, lipid ROS, glutathione peroxidase 4 (GPX4), ChaC glutathione specific gamma-glutamylcyclotransferase 1 (CHAC1), prostaglandin-endoperoxide synthase 2 (PTGS2), solute car- rier family 7 member 11 (SLC7A11), and评估了酰基-COA合成酶长链家族成员4(ACSL4)水平。结果:在癌症组织和细胞中,ARHGAP6显然被下调。ARHGAP6的过表达降低了细胞增殖,细胞死亡升高和脂质ROS,降低了GPX4和SLC7A11,PTGS2,ACSL4和CHAC1增加,并抑制了癌细胞中的RhoA/Rock1和P38 MAPK信号。ARHGAP6敲低与ARHGAP6过表达相反的影响。ARHGAP6 mRNA水平与肿瘤组织中的铁凋亡指标呈正相关。p38 signaling抑制逆转了arhgap6敲低对逆转录病的影响,而rhoa/rock1信号抑制作用损害了arhgap6对p38 mapk信号传导的影响。在小鼠模型中,ARHGAP6以及诱导肌毒死剂RSL3合作的促进性铁氧作用增强并抑制了癌细胞的肿瘤生长。结论:这项研究表明,ARHGAP6通过通过RhoA/Rock1/p38 MAPK信号传递肿瘤来抑制乳腺癌的肿瘤生长。将ARHGAP6与诱导脂肪毒剂诱导剂相结合可能是乳腺癌治疗的有前途的治疗策略。

MKP1通过通过LKB1核保留抑制AMPK活性来促进非酒精性脂肪性肝炎
非酒精性脂肪性肝炎(NASH)是由肝细胞死亡通过caspase 6的激活而触发的,这是由于腺苷一磷酸腺苷(AMP)激活的蛋白激酶-Alpha(AMPKα)活性的降低而引起的。增加的肝细胞膜死亡会促进肝纤维化的炎症。我们表明,在纳什患者和纳什饮食中喂养的雄性小鼠中,核定定位的有丝分裂原活化蛋白激酶(MAPK)磷酸酶-1(MKP1)上调。这项工作的重点是研究MKP1是否以及如何参与NASH的发展。在NASH条件下增加氧化应激,诱导MKP1表达,导致核p38 MAPK去磷酸化并减少肝激酶B1(LKB1)磷酸化,以促进LKB1核出口所需的位点。nash饮食中MKP1的肝缺失喂养雄性小鼠将核LKB1释放到细胞质中,以激活AMPKα并防止肝细胞死亡,炎症和NASH。因此,需要核定定位的MKP1- P38 MAPK-LKB1信号传导才能抑制触发肝细胞死亡和NASH发展的AMPKα。

激酶中的抑制剂诱捕
摘要:最近,我们确定了N- myristoyltransferases(NMTS)中酶抑制的新型机制,我们将其称为“抑制剂捕获”。抑制剂捕获,从而防止其自由解离并导致抑制剂亲和力和效能的显着增加。在这里,我们证明了抑制剂捕获也发生在激酶中。值得注意的是,已经彻底改变了靶向癌症治疗的药物伊马替尼被捕获在ABL激酶的结构中。在p38α激酶中也观察到了这种作用,在p38α激酶中发现抑制剂捕获取决于“魔术”甲基,该甲基稳定蛋白质构象并显着增加了化合物的亲和力。总的来说,这些结果表明,抑制剂诱捕并不是N- myristoyltransferases的独特,因为它也发生在激酶家族中。抑制剂捕获可以增强抑制剂的结合亲和力数千次,并且是在确定药物亲和力和效力中起关键作用的关键机制。

癌症中针对 MAPK 信号传导的综述:药物耐药性和敏感性的机制
通过 3 层激酶级联,从输出到输入信号有负反馈,从而确保对噪声和分级响应的鲁棒性 [2]。MAPK 对各种各样的输入信号作出反应,包括激素、细胞因子和生长因子等生理线索,以及内源性应激和环境信号。因此,传统上将它们分为丝裂原激活 MAPK 和应激激活 MAPK,经典代表有丝裂原反应的 ERK 以及应激反应的 JNK 和 p38。从生理学上讲,这种区别很模糊,因为这三个家族都对各种各样重叠的信号作出反应。MAPK 信号在许多疾病中发生了改变 [3],因此,在过去的二十年里,其激酶成分一直是药物开发的焦点。在癌症和针对 RAS-RAF-MEK-ERK 通路的药物方面取得了最大的进展。人们已经对针对该通路的药物进行了大量的研究,并阐明了敏感性和耐药性的机制。由于研究结果已被广泛综述 [ 4 – 13 ],我们在此仅简要总结一些突出的发现。相反,我们重点讨论 MAPK 信号传导中较少综述的领域及其与耐药性的相关性,即 JNK 和 p38 MAPK 通路,以及与 MAPK 信号传导相关的表观遗传和代谢变化。
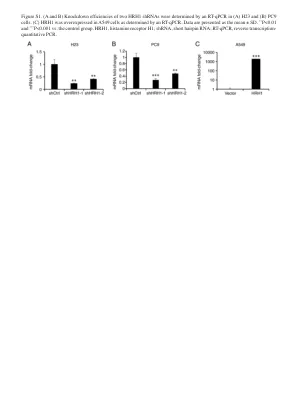
图 S1. (A 和 B) 两个 HRH1 的击倒效率......
图 S3. 使用 Cell Counting Kit-8 检测法评估分别用 10 µM SB203580(p38 抑制剂)、5 µM JNK-in-8(JNK 抑制剂)、30 µM C188(STAT3 抑制剂)或 5 nM OA(PP2A 抑制剂)处理 24 小时的 H23 和 PC9 细胞的存活率。抑制剂处理细胞的存活率以相对于载体处理细胞的百分比表示,载体处理细胞设为 100%。OA,冈田酸。

反对FLR-2糖蛋白激素和DRL的作用...
au:PleaseconfirmthatalleadinglevelsarerepressedCorrected:动物在为增长和繁殖的重要资源提供至关重要的资源之前,整合了发育和营养信号;但是,感知和响应这些输入的途径仍然很少理解。在这里,我们证明了与哺乳动物有丝分裂原激活的蛋白激酶具有相似性的DRL-1和FLR-4在C中保持脂质均匀稳定。秀丽隐章肠。DRL-1和FLR-4在质膜的蛋白质复合物中起作用,以促进发育,因为DRL-1或FLR-4中的突变赋予了缓慢的生长,体积小,体积小和脂质稳态受损。为了确定反对DRL-1/FLR-4的因素,我们对DRL-1突变体表型的抑制剂进行了前遗传筛选,并在FLR-2和FSHR-1中鉴定了突变,该突变分别编码了Folli-Cle刺激激素及其假定的G蛋白蛋白与蛋白质与蛋白质耦合的受体的正交。在没有DRL-1/FLR-4的情况下,神经元FLR-2通过肠道FSHR-1和蛋白激酶A的信号传导来限制生长。此外,我们表明,通过DRL-1和FLR-2的相反信号传导坐在TIR-1寡聚,这调节了下游p38/ pmk-1活性,脂质稳态和发育。最后,我们在肠道中确定了发育转录因子PHA-4/FOXA的令人惊讶的非CA非ca词作用,在该因素限制了响应受损的DRL-1信号传导时,它限制了生长。我们的工作揭示了一个复杂的多组织信号网络,该网络会在p38信号上收敛,以在开发过程中保持体内平衡。