XiaoMi-AI文件搜索系统
World File Search Systempolymerase
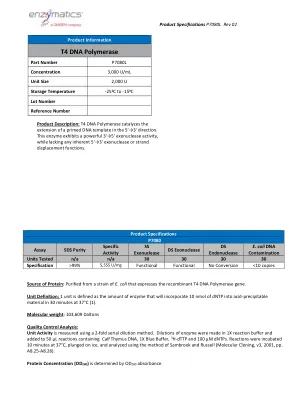
T4 DNA聚合酶
大肠杆菌DNA污染单元已测试了N/A N/A 30 30 30 30规范> 99%5,555 U/mg功能功能性功能性NO转化率<10拷贝<10份蛋白质:从表达重组T4 DNA聚合酶基因的大肠杆菌菌株中纯化。单位定义:1个单位定义为将在37°C下30分钟内将10 nmol的DNTP纳入酸性材料的酶量(1)。分子量:103,609 Daltons质量控制分析:使用2倍连续稀释方法测量单位活动。在1倍反应缓冲液中制成酶的稀释液,并将其添加到含有小腿胸腺DNA,1x蓝色缓冲液,3 H-DTTP和100 µM DNTP的50 µL反应中。在37°C下孵育10分钟,浸入冰上,并使用Sambrook和Russell的方法进行分析(Molecular Cloning,V3,2001,pp。A8.25-A8.26)。蛋白浓度(OD 280)由OD 280吸光度确定。


TAQ DNA聚合酶
TAQ DNA聚合酶是一种重组94 kDa DNA聚合酶,该聚合酶在带有克隆的Thermu Aquaticus DNA聚合酶基因的大肠杆菌菌株中表达。它具有5'-3'聚合酶和外切核酸酶活性,并且没有可检测到的3'-5'外切核酸酶活性。TAQ DNA聚合酶的延伸速率为1-2 kb/ min。此外,它具有3'腺苷酸化活性。因此,PCR产品可直接用于TA-CLONing程序。
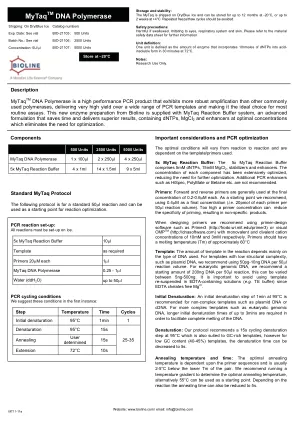
mytaqtm DNA聚合酶
重要的考虑因素和PCR优化,最佳条件将从反应到反应,并取决于所使用的模板/引物。5x mytaq反应缓冲液:5x MyTAQ反应缓冲液包括5mm DNTP,15mm MGCL 2,稳定器和增强剂。每个组件的浓度已得到广泛优化,从而减少了进一步优化的需求。其他PCR增强剂,例如HISPEC,Polymate或Betaine等。。引物:正向和反向引物通常以0.2-0.6M的最终浓度使用。我们建议使用0.4M作为最终浓度(即每50 le反应体积的每个引物的下午20点)。 过高的底漆浓度可以降低启动的特异性,从而导致非特异性产物。 设计底漆时,我们建议使用Primer-Design软件,例如Primer3(http://frodo.wi.mit.edu/primer3)或可单位的10mm和3mm和3mm的阳离子阳离子浓度,或者单位阳离子浓度和分别为10mm和3mm。 引物应具有约60°C模板的熔化温度(TM):反应中的模板量主要取决于所使用的DNA类型。 对于低结构复杂性(例如质粒DNA)的模板,我们建议使用50pg-10ng DNA每50°L反应体积。 对于真核基因组DNA,我们建议每50 l反应的起始量为200ng DNA,这可以在5ng-5ng-500ng之间变化。 避免在含EDTA的解决方案中重新悬浮模板(例如)很重要 te buffer)由于EDTA螯合免费Mg 2+。每50 le反应体积的每个引物的下午20点)。过高的底漆浓度可以降低启动的特异性,从而导致非特异性产物。设计底漆时,我们建议使用Primer-Design软件,例如Primer3(http://frodo.wi.mit.edu/primer3)或可单位的10mm和3mm和3mm的阳离子阳离子浓度,或者单位阳离子浓度和分别为10mm和3mm。引物应具有约60°C模板的熔化温度(TM):反应中的模板量主要取决于所使用的DNA类型。对于低结构复杂性(例如质粒DNA)的模板,我们建议使用50pg-10ng DNA每50°L反应体积。对于真核基因组DNA,我们建议每50 l反应的起始量为200ng DNA,这可以在5ng-5ng-500ng之间变化。避免在含EDTA的解决方案中重新悬浮模板(例如te buffer)由于EDTA螯合免费Mg 2+。初始变性:对于非复合模板,例如质粒DNA或cDNA,建议在95°C下1分钟的初始变性步骤。对于更复杂的模板,例如真核基因组DNA,为了促进DNA的完全融化,需要更长的初始变性时间至3分钟。变性:我们的协议建议在95°C下进行15S循环变性步骤,这也适用于富含GC的模板,但是对于低GC含量(40-45%)模板,可以将变性时间降低到5s。退火温度和时间:最佳退火温度取决于底漆序列,通常比对下的TM低2-5°C。我们建议运行温度梯度以确定最佳退火温度,另外55°C可以用作起点。取决于反应,退火时间也可以减少到5s。


T7 DNA聚合酶 Qiagen验证报告 b'' T7 DNA连接酶 Qiagen®多路复用PCR加套件 T4基因32蛋白
大肠杆菌DNA污染单元测试了N/A N/A 100 100 100规格> 99%13,333 U/mg功能性功能性NO conversion <10份蛋白质来源:重组大肠杆菌菌株,携带毒液T7基因5和E. coli trxa基因。单位定义:1个单位定义为将10 nmol的总DNTPS转换为酸不溶性材料所需的聚合酶量,在37°C下30分钟内。分子量:92.1 KDA质量控制分析:使用2倍连续稀释方法测量单位活动。稀释酶,并将其添加到含有小腿胸腺DNA,1x T7 DNA聚合酶单位表征缓冲液(20 mM Tris-HCl,100 mm KCl,6 mM MGCL,6 mM MGCL 2,6mmmmmgcl 2,0.1 mm EDTA,5 mmβ-MMβ-MERCAPTOETOETHANANOL),3 H-DTT的反应中,3 H-DTT,在37°C下孵育10分钟,浸入冰上,并使用Sambrook和Russell的方法进行分析(6)。蛋白浓度(OD 280)由OD 280吸光度确定。物理纯度,然后进行银色染色检测。通过比较浓缩样品中污染物带的聚集质量与稀释样品中蛋白蛋白蛋白带的质量来评估纯度。单链核酸酶在含有放射性标记的单链DNA底物的50 µL反应中确定,在37°C下孵育4小时4小时。双链外切核酸酶在50 µL反应中确定,该反应含有放射性标记的双链DNA底物和10 µL的酶溶液在37°C下孵育4小时。双链核酸内切酶在50 µL反应中确定,该反应含有0.5 µg质粒DNA和10 µL的酶溶液在37°C下孵育4小时。大肠杆菌16S rDNA的污染是使用5 µL r菌酸溶液的样品变性的样品,并在Taqman QPCR分析中筛选,以使用与16S rRNA locus相应的寡核苷酸引物,使用污染的大肠杆菌Genomic DNA。


PHI29 DNA聚合酶-ABO div>
成分体积底物DNA 10 - 50 ng PHI29 DNA聚合酶反应缓冲液(10x)5 µl DNTPS 0.5 mm抗性的随机六聚体引物5 µM PHI29 DNA聚合酶1 µL(10 U)noclease nuclease nuclease free H 2 O(CAT。编号mb11101)最多50 µL 2。轻轻混合和脉冲。3。在30°C下孵育90分钟。4。在65°C下使酶灭活10分钟。5。继续下游应用程序,例如pCR(用TE缓冲液稀释10-20倍,使用1-3μL稀释的DNA或DNA标记。或者,将放大的DNA存储在-20°C下。避免重复的冻结循环


PrimeSTAR LongSeq DNA 聚合酶
请勿这样使用它。 - 未经 Takara Bio 批准,禁止转售或转让该产品、为转售或转让而修改该产品、或将该产品用于制造商业产品。 ・本宣传册中记载的公司名称及产品名称为各公司的商品名称或注册或未注册的商标,属于各自所有者所有。 ・请访问我们的网站以获取有关许可证等的最新信息。 ・本传单中所列价格为 2025 年 2 月 1 日的建议零售价。价格不含销售税。