XiaoMi-AI文件搜索系统
World File Search Systempotent

抗惊厥药 MK-801 是一种强效的 N-甲基-D-...
摘要 化合物 MK-801{(+)-5-甲基-10,11-二氢-5H-二苯并[a,d]环庚烯-5,10-亚胺马来酸盐} 是一种强效抗惊厥药,口服后有效,其作用机制尚不清楚。我们在大鼠脑膜中检测到了 [3H]MK-801 的高亲和力(Kd = 37.2 ± 2.7 nM)结合位点。这些位点不耐热、具有立体选择性且具有区域特异性,其中海马的位点密度最高,其次是大脑皮层、纹状体和延髓脑桥。小脑中未检测到结合。MK-801 结合位点表现出一种新的药理学特性,因为这些位点上没有一种主要的神经递质候选物活跃。唯一能够竞争 [3H]MK-801 结合位点的化合物是已知能够阻断由 N-甲基-D-天冬氨酸 (N-Me-D-Asp) 受体亚型介导的兴奋性氨基酸反应的物质。这些物质包括分离性麻醉药苯环利定和氯胺酮以及 a 型阿片类药物 N-ailylnormetazocine (SKF 10,047)。使用大鼠皮质切片制剂进行的体外神经生理学研究表明 MK-801 对 N-Me-D-Asp 的去极化反应具有强效、选择性和非竞争性拮抗作用,但对海人酸或奎斯奎特无作用。苯环利定、氯胺酮、SKF 10,047 和 MK-801 对映体作为 N-Me-D-Asp 拮抗剂的效力与其作为 [3H]MK-801 结合抑制剂的效力密切相关 (r = 0.99)。这表明 MK-801 结合位点与 N-Me-D-Asp 受体相关,并解释了 MK-801 作为抗惊厥药的作用机制。

新颖,有效和高度选择性
ATP,三磷酸腺苷;中枢神经系统,中枢神经系统; IC 50,最大抑制浓度的一半; TR-FRET,时间分辨的荧光能传递。 1。 Nassal D等。 前药。 2020; 11:35。 2。 Bezzerides VJ等。 循环。 2019; 140(5):405-419。 3。 Liu Z等。 心律。 2019; 16(7):1080-1088。ATP,三磷酸腺苷;中枢神经系统,中枢神经系统; IC 50,最大抑制浓度的一半; TR-FRET,时间分辨的荧光能传递。1。Nassal D等。前药。2020; 11:35。2。Bezzerides VJ等。循环。2019; 140(5):405-419。 3。 Liu Z等。 心律。 2019; 16(7):1080-1088。2019; 140(5):405-419。3。Liu Z等。 心律。 2019; 16(7):1080-1088。Liu Z等。心律。2019; 16(7):1080-1088。2019; 16(7):1080-1088。


三唑是有效的抗真菌和抗病毒...
引言三唑三唑是五个成员的杂环化合物,具有三个氮(N)原子和两个双键。1,2,4-三唑及其融合的杂环衍生物的化学性质在近几十年来引起了很多关注,它们在合成和生物学上具有重要意义。许多在治疗上有趣的药物候选药物,例如抗真菌药,抗菌。镇痛。抗炎。抗肿瘤。抗病毒。抗惊厥药。抗焦虑。抗组胺药。cns兴奋剂和其他人。包括1,2,3-驱动器部分。[1-8]威胁生命的全身病毒和真菌感染在免疫损害的宿主中越来越普遍,越来越多地研究了三唑衍生物的INHA抑制作用。异尼二氮化物通常抑制INHA。 在FASH系统中的一个重要酶参与分枝杆菌霉菌酸的形成。 通常正在研究1,2,4-三唑的可能的抗病毒和抗肿瘤特性。 这些物质具有1,2,4-三唑残基的示例包括强抗病毒N-核苷利巴韦林和偶氮抗真菌氟康唑。 [9]异尼二氮化物通常抑制INHA。在FASH系统中的一个重要酶参与分枝杆菌霉菌酸的形成。通常正在研究1,2,4-三唑的可能的抗病毒和抗肿瘤特性。这些物质具有1,2,4-三唑残基的示例包括强抗病毒N-核苷利巴韦林和偶氮抗真菌氟康唑。[9]
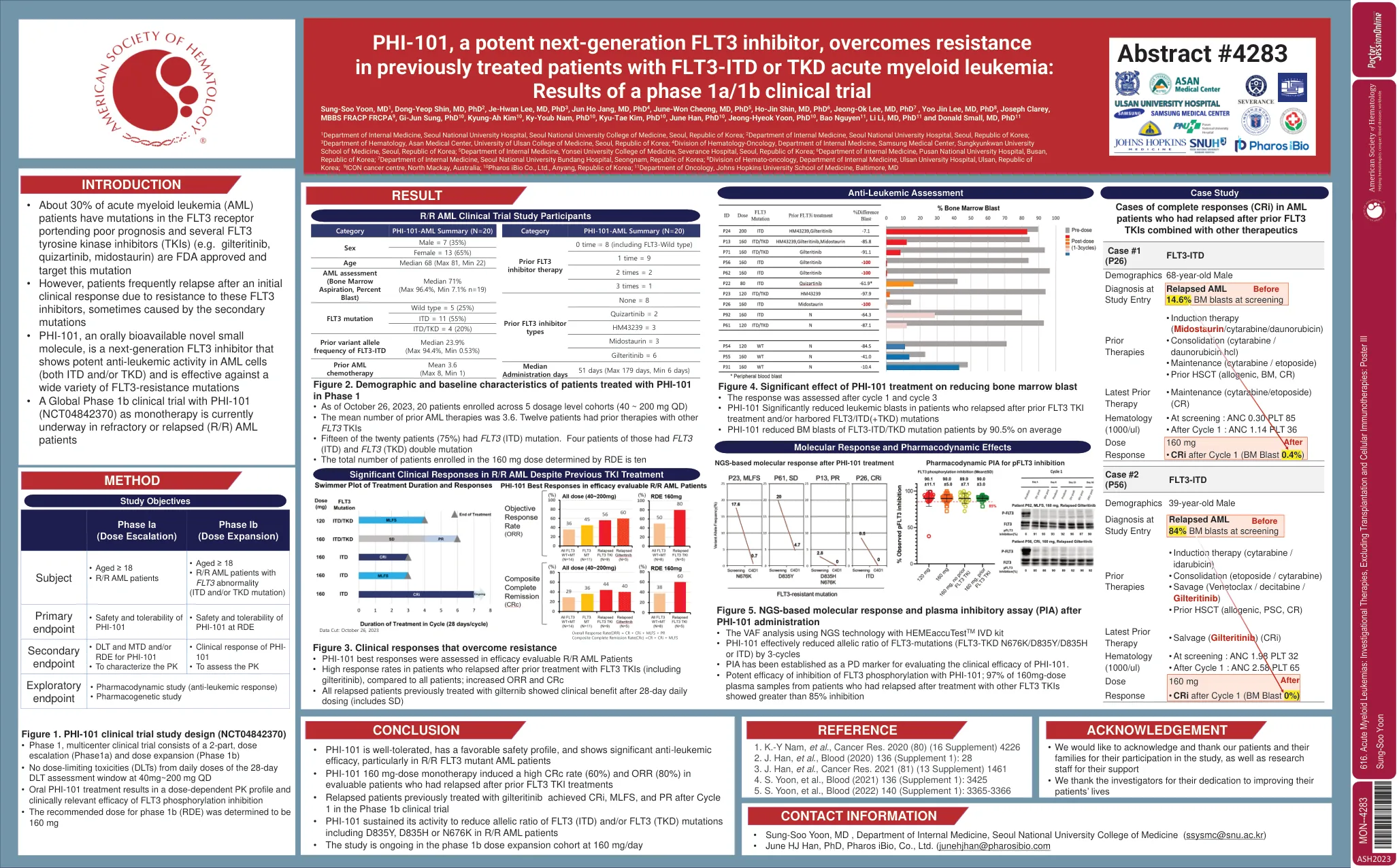
PHI-101,有效的下一代FLT3抑制剂,...
1韩国首尔大学医学院首尔国立大学医院内科,大韩民国共和国; 2大韩民国首尔国立大学医院内科部; 3大韩民国首尔大学医学院阿桑医学中心血液学系; 4大韩民国首尔Sungkyunkwan大学医学院三星医学中心内科中血液学肿瘤学系; 5大韩民国尤斯大学医学院内科医学院内科学系; 6大韩民国釜山普桑国立大学医院内科。 7大韩民国南南北邦医院内科医学系; 8大韩民国乌尔桑大学医院内科医学系血液肿瘤学系; 9澳大利亚北麦凯图标癌症中心; 10号Pharos Ibio Co.,Ltd.,Anyang,大韩民国; 11约翰·霍普金斯大学医学院肿瘤学系,马里兰州巴尔的摩1韩国首尔大学医学院首尔国立大学医院内科,大韩民国共和国; 2大韩民国首尔国立大学医院内科部; 3大韩民国首尔大学医学院阿桑医学中心血液学系; 4大韩民国首尔Sungkyunkwan大学医学院三星医学中心内科中血液学肿瘤学系; 5大韩民国尤斯大学医学院内科医学院内科学系; 6大韩民国釜山普桑国立大学医院内科。 7大韩民国南南北邦医院内科医学系; 8大韩民国乌尔桑大学医院内科医学系血液肿瘤学系; 9澳大利亚北麦凯图标癌症中心; 10号Pharos Ibio Co.,Ltd.,Anyang,大韩民国; 11约翰·霍普金斯大学医学院肿瘤学系,马里兰州巴尔的摩

发现和化学优化有效的双循环(...
。cc-by-nc-nd 4.0国际许可证(未获得同行评审证书)获得的是作者/资助者,他已授予Biorxiv授予Biorxiv的许可,以永久显示预印本。这是该版本的版权所有,该版本于2024年4月9日发布。 https://doi.org/10.1101/2024.03.20.581580 doi:Biorxiv Preprint


NDI-101150是一种有效的高度选择性造血
发表的作者:David.ciccone@nimbustx.com致谢社论援助由Melody Watson,BioScript Group,BioScript Group,Macclesfield,UK提供,Nimbus Therapeutics(Nimbus Discovery Inc.代表Nimbus Saturn Inc.代表Nimbus Saturn Inc.)公开FGB和CL是Nimbus Therapeutics(Nimbus Discovery Inc.代表Nimbus Saturn Inc.)的员工。Alzabin S.等。 癌症免疫学和免疫疗法2010,V59:419-429; 2。 Hernandez S.等。 细胞报告2018。V25:80-94。 3。 Ciccone等人; AACR-NCI-EORTC 2023会议海报C065。 4。 Sommerhalder等。 SITC 2023Alzabin S.等。癌症免疫学和免疫疗法2010,V59:419-429; 2。Hernandez S.等。细胞报告2018。V25:80-94。3。Ciccone等人; AACR-NCI-EORTC 2023会议海报C065。 4。 Sommerhalder等。 SITC 2023Ciccone等人; AACR-NCI-EORTC 2023会议海报C065。4。Sommerhalder等。SITC 2023

开发有效和高度选择性的环氧酮 -
在这里,我们提出了具有低纳摩尔的体外效力的明显基于环氧基酮的蛋白酶体抑制剂,可用于血恶性疟原虫和人类细胞的低细胞毒性。我们的最佳化合物在HEPG2和H460细胞上具有超过2,000倍的红细胞疟原虫的选择性,这在很大程度上是由于P3位置的D-氨基酸的适应D-氨基酸的适应性驱动,并且在P3位置的偏好以及对P1位置的difluorobenzyl群的偏好。我们从恶性疟原虫细胞提取物中分离了蛋白酶体,并确定最好的化合物在抑制恶性疟原虫蛋白酶体的β5亚基方面的有效性更高,与人类成本蛋白酶体的相同亚基相比。这些化合物还显着降低了P. berghei小鼠感染模型中的寄生虫血症,并平均将动物延长6天。当前的环氧基酮抑制剂是口服可生物利用抗疟疾药物的理想起始化合物。